Obesity is a common but complex condition, and many community members and the media hold strong opinions regarding the cause of, and what needs to be done about, obesity. The conventional clinical cut-offs for diagnosis involve calculating body mass index (BMI; body weight in kg/height in m2). A BMI of 25–29.9 kg/m2 is considered overweight, 30–34.9 kg/m2 is obese, and ≥35 kg/m2 is morbid obesity. The Australian Bureau of Statistics (ABS) reports that 63% of Australians were overweight or obese – 27.9% (4.9 million) were obese – in the year ending 2012.1
Adiposity in children is assessed on BMI percentile charts (as is growth) for age and sex, with overweight >85th percentile BMI for age, and obesity >97th percentile BMI for age (World Health Organization criteria).2 There are mixed reports regarding a levelling of obesity prevalence in children in the US and parts of Europe, whereas others report an increase.3
The real morbidity of obesity is related to increased fat tissue content in the body that is adversely affecting health. Assessing fat tissue content most accurately would mean measuring the percentage of true fat in the body, which is most commonly done in a research setting. The metabolic complications of excess fat are influenced by the amount of fat deposited in the central abdominal area. Using a tape measure is a simple clinical way to estimate central fatness by measuring the waist or waist-to-hip ratio. This ratio helps indicate metabolic risk and is also strongly heritable.4
Morbid obesity is a recognised health burden in relation to (among other things) obstructive sleep apnoea, hypertension and cardiovascular disease, musculoskeletal problems, diabetes, social stigma, and depression. However, it is important to remember that possibly up to 30% of people who are obese have no metabolic signs, but still experience the physical burdens of excess weight.5
The difficulty of achieving and maintaining weight loss is well known. A study of the world’s largest primary care electronic practice database examined the probability of a person who is obese losing 5% of total weight (and attaining normal weight) over nine years.6 For morbid obesity, the annual chance of 5% loss was one in eight for men and one in seven for women. For simple obesity, the probability of attaining normal weight was one in 210 for men and one in 124 for women annually, decreasing to one in 1290 for men and one in 677 for women at nine years.
Some of the explanation for the difficulty in achieving and maintaining weight loss lies in the strong heritability of obesity. Classic twin studies have found that a large proportion (40–80%) of variation in body fatness is attributable to genetic differences between individuals.7 The rate of obesity increase in the past three decades varies with race and geography, but is attributable to the effects of rapid environmental change in nutrition and physical activity in genetically predisposed individuals.8 While the ubiquitous availability of tasty, energy-dense foods, and the lessened physical demands of modern life can explain the community weight increase over the past three decades, a heightened susceptibility of certain members of the community to this obesogenic environment suggests a strong influence of underlying genetic factors.
Regulation of body composition
It is important to understand that it is regulation of body composition itself that appears to be under genetic control. The classic adoption studies of Stunkard originally found that adopted children resembled (in thinness and fatness) their genetic parents (whom they had never met) more than the adoptive parents they had lived with. The reported heritability of BMI in twin studies was around 70–80%.9 In our own twin studies, we found separate heritable influences affecting whether fat is deposited centrally or peripherally.4 Claude Bouchard, in elegant studies also in monozygotic and dizygotic twins, confirmed such twin pair resemblances in the amount of weight gained or lost when challenged by diet or overfeeding.10
The now famous discovery that severe obesity in a child is due to defective production of the hormone leptin by the leptin gene in fat cells, with successful treatment by recombinant human leptin, launched the current era of rare genetic mutations discovery. Lack of leptin in an individual produces extreme hyperphagia, hypogonadism and reduced sympathetic tone, which are all reversed by the replacement of leptin.11 Through the study of extreme obesity cases and their families with clinical presentations of morbid obesity (usually presenting in children), identification of many rare genetic defects has been possible. This has provided valuable insight into the mechanisms controlling appetite and energy balance involved in the overall pathophysiology of obesity.
Leptin, produced by fat cells, signals to the hypothalamus via the leptin receptor (whose gene is also reported to have rare mutations) that there are ‘enough’ nutrients. Leptin stimulates expression of pro-opiomelanocortin (POMC) in the arcuate nucleus in the hypothalamus, which activates multiple receptors in the central nervous system (CNS) while inhibiting appetite-related pathways (Figure 1). The gene product of POMC is split to produce alpha-melanocyte-stimulating hormone (á-MSH), which binds to the melanocortin 4 receptor (MC4R), which then propagates the anorexic message from leptin downstream to the brain and onwards. In cases of more severe obesity presenting in early life, rare mutations in a number of molecules that modulate energy balance via the hypothalamic leptin/melanocortin pathway have now been described. This includes POMC mutations present with adrenocorticotropic hormone (ACTH)-dependent cortisol deficiency; patients with this mutation have reddish hair, lack skin pigment and develop severe obesity early in life. Hyperphagia with an MC4R gene defect is less severe than with leptin, and is a relatively common form of severe genetic obesity (up to 5% of severe, early onset obesity), reported as one in 1000 in the UK.12 Children with MC4R deficiency have increased growth and final height, and are hyperphagic and hyperinsulinaemic; however, they are not as ravenous as those with a leptin deficiency and have lower blood pressures than patients who are equivalently obese, because of less sympathetic nervous system involvement. A number of drugs that target MC4R are under development for future pharmacological treatment.
Melanocortin peptides are processed by enzymes, including prohormone convertase 1 (PC1), which converts POMC into ACTH. Genetic defects reported in PC1 are also known to cause early-onset obesity with hyperphagia.3 Polymorphisms in two genes found downstream of MC4R (BDNF1 and SIM1) in the hypothalamus have also been linked with obesity. Subjects with SIM1 mutations share features with those with MC4R deficiency, including hyperphagia and autonomic dysfunction, and variable developmental delay.3
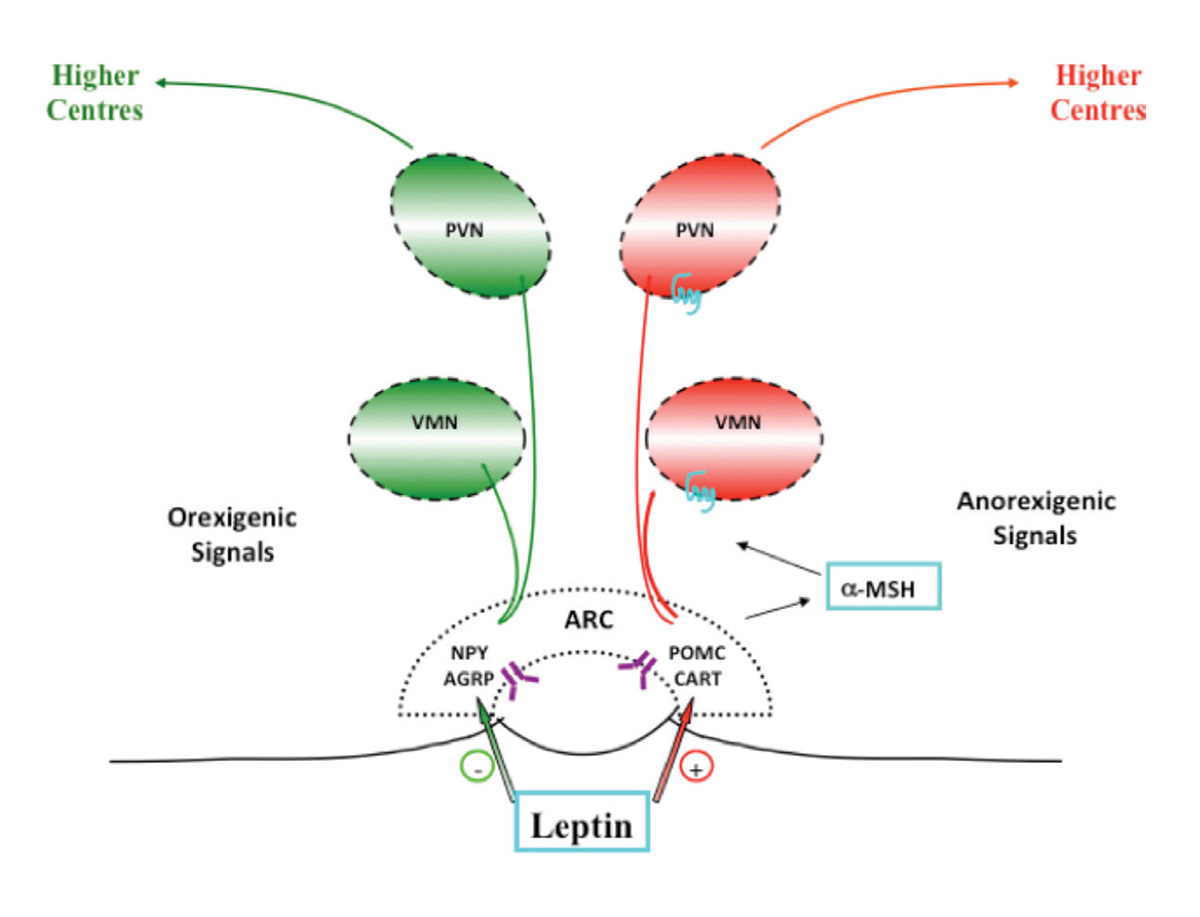
Figure 1. Leptin pathway
ARC, arcuate nucleus; AGRP, agouti-related protein, CART, cocaine- and amphetamine-regulated transcript; α-MSH, alpha-melanocyte-stimulating hormone;NPY, neuropeptide Y; POMC, pro-opiomelanocortin; PVN, paraventricular nucleus; VMN, ventromedial nucleus
Genome-wide association study
In the search for influence of common genes on obesity, large numbers of people have to be studied; therefore, BMI is commonly used (as it is easily measured in large numbers, despite the fact that percentage of fat reflects true fatness far more accurately). The genome-wide association study (GWAS), where populations that are obese are compared with large numbers of lean people, have explained less than 5% of the heritability of obesity. This is despite GWAS having identified almost 100 common single-nucleotide polymorphisms (SNPs) in genes over the years. It is more likely that rare genetic variants, which outnumber the common ones in the human genome (the ones being studied in GWAS), actually explain more of the heritability of human obesity and can be more readily identified at the upper end of the obesity spectrum, where the major obesity increase is occurring in our strongly obesogenic environment.13
The most common GWAS links to obesity were in the FTO gene (on 16p11.2).11 Obesity-associated sequences within FTO appear functionally connected through a non-coding ribonucleic acid (ncRNA) to an increased expression of IRX3, an adipose tissue gene that has been shown to have an effect of browning white fat.12 This obesity FTO SNP can have the obesity effect attenuated by physical exercise and possibly by dietary interventions,14 giving hope that some people may one day have suitable lifestyle treatments prescribed for specific genetic defects.
Syndromic obesity
Obesity is sometimes part of a rare but recognised syndrome characterised by cognitive delay, dysmorphic features and organ-specific abnormalities. These can be identified in children:
- Prader–Willi syndrome, an imprinting disorder, is the most common, occurring at a rate of one in 15,000. Babies with this syndrome are born ‘floppy’, have weak muscles and up-slanted eyes, and feed poorly. Over the first two years, the infant slowly develops a ravenous appetite, and has cognitive delay and behavioural issues. It is an imprinting defect, in the region of chromosome 15q11–13.
- A variant of the Fragile X syndrome has also been described that has features of Prader–Willi syndrome, with severe obesity developing early.12
- Bardet−Biedel syndrome is an autosomal recessive disorder of ciliary function characterised by obesity, cognitive delay, polydactyly and retinitis pigmentosa, renal dysfunction and hypogonadism, sometimes resulting in diabetes. It has so far been mapped to 19 different genes.
- Albright’s hereditary osteodystrophy (pseudohypoparathyroidism) is associated with hyperphagia, obesity, short stature, round face, skeletal anomalies and cognitive delay. The GNAS gene mutation results in resistance to hormones such as parathormone, thyroid hormone and gonadotrophins.
- WAGR syndrome (11p14 deletions leading to brain-derived neurotrophic factor haploinsufficiency) is characterised by aniridia, obesity, growth retardation, cognitive delay and genitourinary deformities.
Pigeyre, Yazdi, Kaur and Meyre list 10 forms of syndromic obesity in a review of recent progress in understanding the genetic basis of obesity.12
Copy-number variations (CNVs) in the genome have also been described in association with obesity. CNVs are segments of the genome that differ in the number of diploid copies carried by healthy individuals. About 5% of healthy people have common varieties of CNV, and 1% have rare varieties. Common CNVs include chromosome 10q11.22, a region associated with the pancreatic polypeptide receptor, where low CNV was recently reported to be associated with high BMI, and also on chromosome 11q11 around the olfactory receptor genes with early onset extreme obesity.11
Implications for the future
For the moment, the notion of ‘tailored genomic medicine’ is still a promise. As we detect each specific genetic defect, we may be able to tailor treatment for the patient by replacing a gene product, altering a diet or tailoring a drug specifically to improve the defect. So far, this has been achieved clinically in patients with leptin deficiency who are treated with recombinant leptin replacement.
Currently, whole-exome sequencing (WES) of the genome of cohorts with severe obesity (child or adult) and those at the extremes of the BMI classification is underway. It provides a ‘most complete’ view of all genomic variations and is likely to identify new genes (and variants) whose function will then need to be tested further experimentally. We predict that future studies, using more accurate phenotyping, WES and more accurate physiological measures, will progressively reveal more about as yet undiscovered rare, dominant (or additive) genetic determinants of obesity.14
The identification of many factors involved in weight control has already helped shed light on the pathways and mechanisms by which obesity develops. Novel mutations continue to be reported in severe obesity that could enable carriers of such mutations to have more specific therapy. So far, this is in the research stage, but some progress has been made:
- Successful testing of an MC4R partial agonist for appetite normalisation and inhibition of compulsive eating has been reported in two patients with the genetic defect.15
- People who are obese often think they have a ‘slow metabolism’, but this appears to be rare in humans. However, there is a recent report of a mutation in a gene called KSR2, which codes for a scaffolding protein that appears to be associated with a lower metabolic rate. A promising early report suggests that metformin may be beneficial in reversing the defect in KSR2 carriers, but more studies are needed.11
Key points
Some clinical messages are clear from the research so far.
- The knowledge that a person has a strong family history of obesity and/or type 2 diabetes mellitus is an alert that they may be vulnerable to weight gain. Early lifestyle advice (including regular activity) may prevent or delay the onset of lifetime weight problems.16
- A no blame/no guilt approach should be adopted when treating patients with morbid obesity, as we are treating a strongly heritable disorder for which patients often blame themselves lifelong. Physicians should not add more blame or guilt.
- Early-onset hyperphagia and obesity in children should be regarded as a flag to consider specialist referral, especially for those with a positive family history or syndromic features, such as cognitive problems, growth disorders or developmental delay. Early treatment can prevent severe obesity and help with family counselling.
- Patients are interested in knowing about any predisposing genetic findings. Preliminary evidence suggests that receiving genetic test feedback (if available) can relieve stigma and self‑blame related to weight gain, and could increase readiness to control weight, with evidence of weight benefits in a randomised trial.17
- While there is not yet sufficient evidence, early findings indicate that genetic differences may help determine which approach is more effective (or at least attractive) for any individual. Comparison studies of weight-loss diets (eg low carb, low fat) have found that the ‘successful’ diet is the one the patient prefers. Current known genetic ‘carriers’ (FTO gene) are reported to lose more through diet and lifestyle interventions than non‑carriers, which may encourage lifestyle efforts in those who are otherwise discouraged by having obesity ‘in the family’.18
- Bariatric surgery currently remains the most successful therapy for morbid obesity (with long-term follow-up initially in the expert surgical centre, essential for nutritional adequacy) until we have evidence from long-term trials of novel drug therapy for severe obesity.
Author
Lesley V Campbell AM, MBBS, FRACP, FRCP, Professor of Medicine, UNSW; Laboratory Co-Head, Clinical Diabetes, Appetite and Metabolism, Diabetes and Metabolism Division, Garvan Institute of Medical Research, Sydney, NSW; and Senior Endocrinologist, Department of Endocrinology, St Vincent’s Hospital Sydney, Darlinghurst, NSW. l.campbell@garvan.org.au
Competing interests: None.
Provenance and peer review: Commissioned, externally peer reviewed.