Case
A retired engineer, 67 years of age, visited his general practitioner (GP), complaining of increasing breathlessness with exertion over 12 months. He also noticed a dry cough and his fingernails had become increasingly rounded. Further history revealed that he was an ex-smoker with a 15 pack-year history, had limited past exposure to asbestos, no significant environmental exposures to mould, birds, dust or other organic materials, and was only taking irbesartan (for hypertension). His resting oxyhaemoglobin saturation was 96%, which dropped to 87% with sit-to-stand exercise. He had marked digital clubbing, but no features to suggest an autoimmune connective disease process. Fine velcro-like crepitations were heard bi-basally on chest auscultation. Cardiovascular findings were normal and there was no evidence of pulmonary hypertension or heart failure. Spirometry suggested a restrictive lung disease with moderate reductions in forced expiratory volume in 1 second (FEV1, 1.8 L, 69%) and forced vital capacity (FVC; 2.2 L, 67%), but a preserved spirometric ratio (88%). There was no improvement in measurements with an inhaled bronchodilator. The GP suspected the patient had interstitial lung disease.
Interstitial lung disease (ILD) is a heterogeneous group of disorders that are characterised by varying degrees of fibrosis and inflammation of lung parenchyma. This review will discuss the updated ILD classification scheme and summarise recent landmark advances in the field (including two new, effective antifibrotic agents) which have resulted in changing perspectives in the management of ILD.
Common clinical features in interstitial lung disease
Patients with ILD often report progressive shortness of breath, exercise intolerance and a pervasive dry cough. Fine crepitations may be appreciated on chest auscultation. Signs of pulmonary hypertension and right heart failure may also be present, particularly in advanced disease. Peripheral features of connective tissue disease should be considered in patients with ILD, as these are important clues to the underlying aetiology. Oxygen desaturation commonly occurs during exertion and is associated with poorer long-term survival.1,2 Resting hypoxia is a sign of advanced disease. In contrast to chronic obstructive pulmonary disease (COPD), carbon dioxide retention is not typically observed in ILD until the terminal stages.
Important investigations in diagnosing ILD
Establishing an accurate ILD diagnosis is critical, as different disease subtypes carry distinct prognoses and require tailored management strategies. Baseline and more detailed investigations are shown in Table 1. A preliminary plain chest X-ray is usually performed as a baseline measure, although it is a non-specific and insensitive test in patients with ILD. High-resolution computed tomography (HRCT) scanning has emerged as an invaluable tool for the assessment of patients with ILD. Supine and prone images should be obtained to remove artefacts in gravity-dependent areas. Comparing inspiratory and expiratory series allows for the detection of gas trapping or ‘mosaicism’, which is a feature in some ILD subtypes.3 Lung function and exercise testing (usually with a simple 6-minute corridor walk test) are important in establishing the functional impact or severity of the disease. Detailed serology is important in identifying underlying connective tissue disease. More comprehensive testing (eg bronchoscopy and surgical lung biopsy) may provide further diagnostic information, but is not necessary in all cases. A ‘clinical–radiological–pathological’ diagnosis is most accurately established within multidisciplinary discussion, where ILD physicians, radiologists and pathologists collaborate dynamically. The multidisciplinary discussion is considered the gold standard for determining a diagnosis of ILD, minimises observer bias and enhances diagnostic confidence.4,5
Table 1. Interstitial lung disease investigations
|
|
|
Investigation
|
Possible findings
|
|---|
|
Routine at baseline and follow up
|
|
|
|
|
- Nodules
- Cysts
- Ground glass change
- Honeycomb change
- Traction bronchiectasis
- Intralobular septal thickening
|
- Pulse oximetry/ arterial blood gas
|
|
- Connective tissue disease serology
|
- Positive autoantibodies (eg ANA, ENA, RF, myositis antibodies, ANCA)
|
- Lung function tests (spirometry, lung volumes, DLCO)
|
- Low FEV1, FVC
- Normal or high FEV1/FVC ratio
- Reduced lung volumes
- Reduced DLCO
|
|
|
- Reduced walk distance
- Oxygen desaturation
|
|
Occasional
|
|
- Variable, frequently normal
- May have elevated neutrophils, eosinophils and/or lymphocytes
|
|
|
- Variable and specific for diagnosis
|
|
|
- Pulmonary hypertension
- Right ventricular dysfunction
|
|
|
- Confirmation of pulmonary hypertension
|
|
|
- Nocturnal hypoxia
- Obstructive sleep apnoea
|
|
HRCT, high-resolution computed tomography; SpO2, oxyhaemoglobin saturation; PaO2, partial pressure of arterial oxygen; ANA, antinuclear antibodies;
ENA, extractable nuclear antigens; RF, rheumatoid factor; ANCA, anti-neutrophil cytoplasmic antibody; FEV1, forced expiratory volume in 1 second;
FVC, forced vital capacity; DLCO, diffusing capacity of the lung for carbon monoxide
|
ILD classification scheme
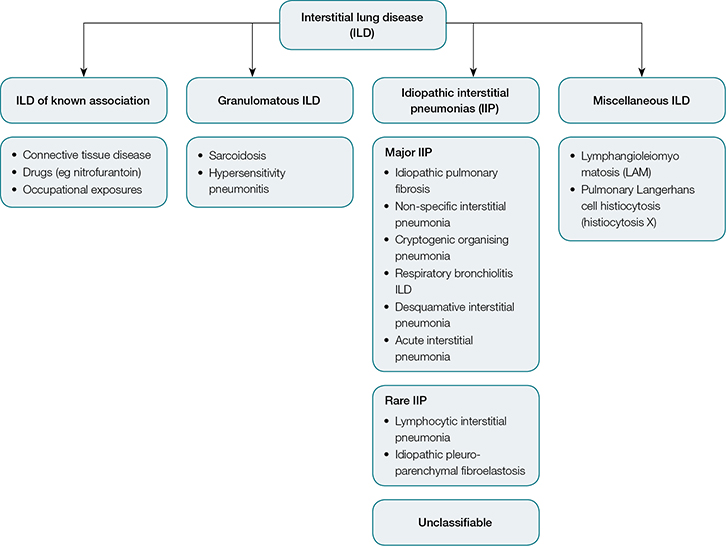 |
| Figure 1. Interstitial lung disease classification5,8 |
Establishing an accurate diagnosis of ILD can be challenging for clinicians as there are more than 200 different subtypes. Over the past decade, ILDs have been reclassified in comprehensive international consensus statements.5–8 The major subgroups of ILD (Figure 1) are broadly defined as:
- idiopathic interstitial pneumonias (IIPs)
- granulomatous ILD (eg sarcoidosis, hypersensitivity pneumonitis)
- ILD with known associations (eg occupational and drug exposures, connective tissue diseases)
- miscellaneous ILD (eg pulmonary Langerhans cell histiocytosis [PLCH]).
Determination of the disease subtype requires consideration of the patient’s history of exposures, specific clinical features, serology, radiological pattern and, in some cases, lung biopsy during multidisciplinary discussions. Surgical lung biopsy carries an inherent risk of morbidity and mortality and only a fraction of patients are deemed suitable.9
Idiopathic interstitial pneumonias
This group of diseases includes:
- chronic fibrosing IIP (idiopathic pulmonary fibrosis [IPF] and non-specific interstitial pneumonia [NSIP])
- smoking-related IIP (desquamative interstitial pneumonia [DIP] and respiratory bronchiolitis ILD [RBILD])
- acute/subacute IIP (cryptogenic organising pneumonia [previously known as bronchiolitis obliterans with organising pneumonia, BOOP] and acute interstitial pneumonia [AIP]).
Revised guidelines in 2013 included idiopathic pleuroparenchymal fibroelastosis (IPPFE), a newly described rare entity, and ‘unclassifiable ILD’ for cases where diagnosis cannot be confidently established.5 Approximately 10% of patients referred to specialist ILD clinics will fall into this category, primarily because of the inability to undergo surgical lung biopsy.10,11 This proportion is likely to be even higher outside tertiary referral centres.
Idiopathic pulmonary fibrosis
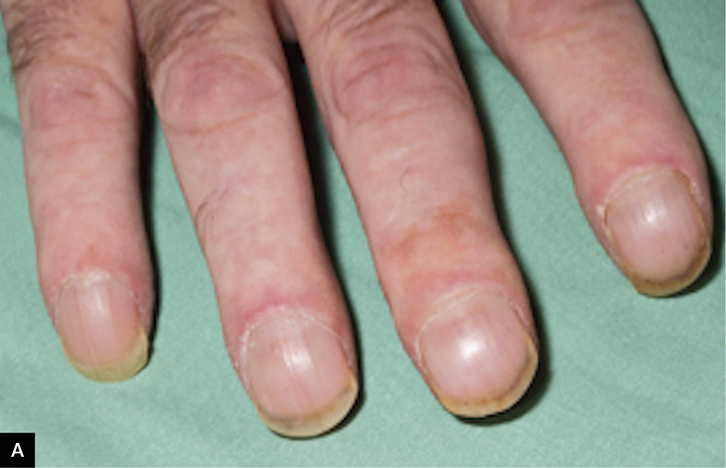 |
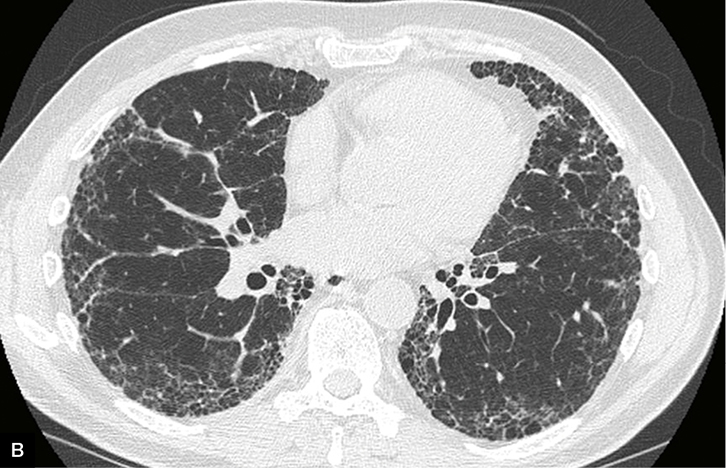 |
Figure 2. Clinical features in idiopathic pulmonary fibrosis
A. Digital clubbing
B. Usual interstitial pneumonia pattern on high-resolution computed tomography |
IPF is the most common IIP subgroup and is thought to be a disease of ageing, as most cases develop in people aged 60 years and older. The estimated prevalence is 2–29 cases per 100,000 in the general population, but higher in older age groups.6 There is a male predominance, and a history of smoking is frequently found. Other potential risk factors include gastro-oesophageal reflux disease (GORD) and specific genetic mutations.6 IPF appears to arise from a process of primary fibrosis, distinguishing it from other IIPs and many other ILD subtypes, which are characterised by inflammation of lung tissue prior to the development of interstitial fibrosis. Digital clubbing may be present, but can also develop in other fibrotic lung diseases as they progress (Figure 2a). The clinical vignette in the introduction describes a typical patient with IPF, but a number of differential diagnoses would still need to be considered.
In approximately two-thirds of patients with IPF, a confident diagnosis can be established when other known causes of pulmonary fibrosis are excluded (through careful history and serological evaluation), and typical radiological features are present.6 The characteristic ‘usual interstitial pneumonia’ (UIP) pattern on HRCT seen in IPF includes basal, sub-pleural reticular abnormalities, and honeycombing and traction bronchiectasis (Figure 2b). A lung biopsy revealing histological UIP is required to make a definite diagnosis in the remainder of cases where radiological findings are suggestive but not definite. IPF is associated with poor prognosis, with a 2–3-year median survival from diagnosis.6 On the other hand, patients with NSIP, have a significantly more favourable course, with median survival greater than 9 years.12
Cryptogenic organising pneumonia
Cryptogenic organising pneumonia (COP) also warrants special mention as a common IIP. This disease of uncertain aetiology may present similarly to infective pneumonia, with single or multifocal air space opacities, in association with malaise, fevers and cough. Diagnosis is frequently delayed. However, prognosis is generally excellent with a good response to systemic corticosteroids.13 The organising pneumonia pattern is also seen in association with certain connective tissue diseases and secondary to some medications.
Granulomatous ILD
Pulmonary sarcoidosis and hypersensitivity pneumonitis are two important and distinct disease processes within this major ILD subgroup. Sarcoidosis is a multi-system disease that results in pulmonary fibrosis in up to 20% of patients.14 Little is known about the aetiology of this common condition. However, well-formed granulomas, the characteristic pathological lesion, are thought to form in response to a variety of environmental stimuli (eg infection, inorganic particles and certain chemicals).15 Important differential diagnoses include granulomatous lung disease from specific infections (eg Mycobacterium tuberculosis) and medications (eg methotrexate).15 The majority of cases of pulmonary sarcoidosis will remain stable or regress, and will not require specific therapy. Immunosuppressive therapy may be instituted where there is progressive lung fibrosis, or when other organs are involved (including cardiac and ocular sarcoid).
Hypersensitivity pneumonitis (also known as extrinsic allergic alveolitis) is another granulomatous lung disease that shares overlapping clinical, radiological and histological features with IPF and fibrotic NSIP.5 This is often chronic, with insidiously progressive fibrosis, although it may present in a fulminant or acute stage. This disease develops when susceptible individuals become sensitised to environmental antigens (eg from birds, mouldy hay and potting mix) or certain drugs. The inciting factor is obscure in 30% of patients, even after taking a detailed history.16
ILD of unknown association
Connective tissue disease associated ILD
ILD is a well-described manifestation of certain connective tissue diseases (CTD). This includes scleroderma, systemic lupus erythematosus, rheumatoid arthritis, dermatopolymyositis and mixed connective tissue disease. Immune-mediated inflammation of the lung interstitium can occur, sometimes progressing to irreversible fibrosis if it is left untreated. In some cases, the ILD may be the first manifestation of a wider systemic disease, pre-dating other symptoms by months and even years. In a subset, the pulmonary disease may be the primary feature of the autoimmune process.17 A detailed panel of autoimmune serology (Table 1) is important in all patients with newly diagnosed ILD.
Cigarette smoking and ILD
RBILD, DIP and PLCH are specific ILDs that have a strong causal association with cigarette smoking.18,19 Smoking is likely to induce an immune response in certain individuals, leading to the development of these diseases. Smoking cessation may often be adequate to bring about clinical stability or even remission.20 Previous or current smoking also increases the risk of developing IPF and rheumatoid-associated ILD. However, the temporal relationship is less clear than with RBILD, DIP and PLCH.21 Recently, the co-existence of pulmonary fibrosis and emphysema has been defined as a syndrome (combined pulmonary fibrosis and emphysema [CPFE]), characterised by a high prevalence of pulmonary hypertension (PH) and generally poor prognosis.22
Management of ILD
Accurate diagnosis within an multidisciplinary discussion is essential prior to establishing a management plan. Disease behaviour should also be identified, with frequent revision at follow-up assessments. This is particularly important for patients with ‘unclassifiable disease’ during multidisciplinary discussions. Immediate removal of the exposure is crucial in patients in whom exogenous agents are suspected of causing the ILD. Careful questioning of current or recent contact with environmental mould, birds, tobacco smoke and specific medications is important. In many cases, simply removing the inciting agent may be sufficient to bring about disease regression without the need for immune suppression.
Idiopathic pulmonary fibrosis
Until recently, there were no specific treatment options for IPF other than lung transplantation.23 The previous widespread practice of using immunosuppressive therapy in IPF was found to be ineffective and associated with worse outcomes.24 In 2014, two antifibrotic agents (nintedanib and pirfenidone) were found to reduce the decline in lung function over 12 months (Table 2).25,26 Pirfenidone also seems to confer a small mortality benefit. These antifibrotic therapies have recently been licenced for prescription in patients with IPF in the US and Europe and are currently being prescribed in certain specialist centres in Australia. Eligible patients need to fulfil certain trial criteria
(ie 40 years or older, definite IPF at multidisciplinary discussions, and with mild-to-moderate severity). Other antifibrotic agents are being tested in clinical trials and it is certain that IPF management strategies will continue to evolve.
Table 2. Results of recent clinical trials in idiopathic pulmonary fibrosis
|
|
Trial
|
Drug
|
Number of patients
|
Inclusion
|
Findings
|
|---|
|
INPULSIS I
INPULSIS II23
(Phase III)
|
Nintedanib
|
515
555
|
- Age >40 years
- Mild-to-moderate disease (FVC >50%, DLCO 30–79%)
- FEV1/FVC >0.7
|
- 50% reduction in annual rate of decline in FVC
- Increased time to acute exacerbation
- Improved quality of life (INPULSIS I only)
|
|
ASCEND24
(Phase III)
|
Pirfenidone
|
555
|
- Age 40–80 years
- Moderate disease (FVC 50–90%, DLCO 30–90%)
- FEV1/FVC >0.8
|
- 45.1% reduction in change FVC over 52 weeks
- Reduced decline in 6MWD
- Improved progression-free survival at 52 weeks
- Pooled data with previous trials: reduced all-cause and IPF-related mortality
|
|
FVC, forced vital capacity; DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 second; 6MWD, six-minute walk distance; IPF, idiopathic pulmonary fibrosis
|
Inflammatory ILD
In contrast to IPF, treatment for other ILD (including NSIP, CTD-associated ILD and hypersensitivity pneumonitis) is targeted at reducing inflammation, to prevent fibrosis and maintain function. Corticosteroid-based regimens are standard, often with addition of steroid-sparing agents including azathioprine, mycophenolate and cyclophosphamide. In cases where treatment is delayed or ineffective, patients may require referral to a transplant centre or palliative care services, depending on their age and burden of disease.
Transplant referral
Lung transplantation is a potentially curative option for a select group of ILD sufferers. Timely transplant referral is critical, particularly for younger patients with treatment-refractory, aggressive fibrosis. The rate of decline in FVC and diffusing capacity of the lungs for carbon monoxide (DLCO) is an important predictor of the requirement for transplantation in patients with IPF.6
Managing symptoms and comorbidities
Early recognition and treatment of infections, and vaccinations, supplemental oxygen and treatment of comorbid conditions may help to preserve quality of life in many patients with ILD. Some comorbidities are particularly common in ILD and require targeted therapy (Table 3).
Table 3. Common comorbidities in interstitial lung disease
|
|
Comorbidity
|
Potential implications
|
Management
|
|---|
|
Gastro-oesophageal reflux disease
|
Lung injury, acceleration of lung fibrosis
|
- Proton pump inhibitor
- H2-receptor antagonist
- Prokinetic agents (mainly scleroderma)
- Surgery in selected cases
|
|
Osteoporosis*
|
Vertebral and rib fractures can further restrict breathing, as well as impact on quality of life
|
- Vitamin D repletion
- Surveillance bone mineral densitometry
- Bisphosphonates
- Endocrinologist referral in some cases
|
|
Infections
|
Accelerate decline in lung function
|
- Influenza and pneumococcal vaccinations
- Minimise sick contacts
- Timely antibiotic therapy
|
|
Sleep-disordered breathing
|
May contribute to pulmonary hypertension; sleep fragmentation can affect quality of life
|
- Nocturnal oxygen
- Continuous positive airway pressure
|
|
Pulmonary hypertension
|
Increased mortality
|
- Supplemental oxygen
- Diuretics (for right heart failure)
- Vasodilator therapy in selected cases
|
|
*Often accelerated with corticosteroid use
|
Gastro-oesophageal reflux disease
Uncontrolled reflux can cause direct lung injury and is thought to be an important contributing factor to the pathogenesis of IPF.6 Although guidelines advocate GORD treatment in IPF patients, even when asymptomatic of reflux, it is not clear if this has any impact on the clinical course of the disease. Oesophageal dysmotility is a key feature in scleroderma, and patients often need proton pump inhibitor and prokinetic therapies.
Pulmonary hypertension
Pulmonary hypertension is a common manifestation in many patients, conferring poorer prognosis.27–29 It often occurs late in the disease course, but may also occur at an earlier stage, particularly in connective tissue disease. Vasodilator therapy may be trialled in some cases, and such patients should be managed in specialised pulmonary hypertension clinics.
Obstructive sleep apnoea
Nocturnal hypoxia and obstructive sleep apnoea are highly prevalent in ILD populations. Supplemental oxygen and continuous positive airway pressure may be prescribed, although evidence for their benefit is currently lacking.30
Summary
The patient described in the case presentation requires careful evaluation to determine his ILD diagnosis and best course of management. There is a strong imperative to accurately identify the underlying disease to ensure optimal outcomes. With increasing understanding of the pathological processes, new, targeted treatment options are being developed. Symptom burden and comorbid conditions have a substantial impact on quality of life in patients with ILD, and also warrant careful attention.
Authors
Lauren Troy MBBS, BMedSci, FRACP, Respiratory Staff Specialist, Department of Respiratory Medicine, Royal Prince Alfred Hospital, Camperdown, NSW; Sydney Medical School, University of Sydney, NSW
Tamera Corte MBBS, BSc (Med), FRACP, PhD, Associate Professor, Respiratory Staff Specialist, Department of Respiratory Medicine, Royal Prince Alfred Hospital, Camperdown, NSW; Sydney Medical School, University of Sydney, NSW. tameracorte@mac.com
Competing interests: Tamera Corte is paid as an advisory board member of and for consultancy to Astra Zeneca and Boehringer Ingleheim. She has received payment for speaking engagements by the latter. She has also had expenses covered by GSK, Boehringer Ingelheim and Actelion. Lauren Troy has had expenses covered by Bayer, Intermune and Gilead. The authors’ institution has also received grants from Boehringer Ingeleheim, Intermune, Bayer and Actelion.
Provenance and peer review: Commissioned, externally peer reviewed.