Even healthy children have infections more frequently than adults, which can raise parental concern about an inadequate immune system. The clinician dealing with these concerns needs to know what is normal in terms of frequency and severity of childhood infections, and to be on the lookout for signs that may suggest immunodeficiency diseases. These may be primary immunodeficiencies (PIDs), which include a group of inherited diseases that result in a decreased ability of the immune system to respond effectively to pathogens. Secondary or acquired immunodeficiencies (SIDs) may be caused by drug toxicities, infection or malignancy. Recognising an immunodeficient state in children can be a challenge and this review will direct the reader towards some of the more useful clinical signs and basic diagnostic investigations, with an emphasis on PIDs.
Primary immunodeficiencies
PID involves an infectious predisposition associated with a deficiency of certain immune components. There are more than 250 characterised PIDs affecting an estimated 1 in 1200 live births.1 The infectious predisposition (eg viral, bacterial or fungal) differs, depending on which gene or genes are involved, with more severe deficiencies presenting early in infancy. Early-onset PIDs often have a Mendelian inheritance pattern and many are X-linked, occurring more commonly in boys.
PIDs are often divided into groups. One classification includes T-cell/cell-mediated or combined deficiencies, B-cell/antibody deficiencies, deficiencies affecting phagocytes, deficiencies of cytokine or complement pathways, and syndromic immunodeficiencies where the infectious predisposition is accompanied by other syndromic features.2
Secondary immunodeficiencies
Compared with PIDs, SIDs are much more common. The SID that comes most readily to mind for most clinicians is acquired immunodeficiency syndrome (AIDS), which results from the destruction of CD4+ T-cells by human immunodeficiency virus (HIV), with the consequent acquisition of opportunistic infections. However, HIV infection in childhood is now rare in Australia; with the excellent antenatal care, only about 100 cases have been reported in Australia since the start of the epidemic. This disease is most likely to occur when there has been inadequate pregnancy screening, for example, when the mother arrives late in pregnancy from an HIV-endemic country.3
Drugs are by far a more important cause of SID, particularly immunosuppressive regimes used for cancer and autoimmune diseases. At its simplest, the risk of infection depends on the degree of immunosuppression; anti-rheumatic treatments generally confer a lower risk of infection than chemotherapy for cancer or haematopoietic stem cell transplant. However, many of the regimens possess very specific risks, such as the risk of tuberculosis and Legionella pneumonia associated with TNF-á blockers.4,5 Malnutrition is another important cause of SID, as are trauma/surgery, indwelling lines (which disrupt physical barriers), splenectomy, chronic diseases such as renal disease, and the extremes of age.
How do we recognise immunodeficiencies?
Immunodeficiencies tend to be associated with either excessively frequent or severe infections, sometimes with organisms of low pathogenicity. It is important, therefore, to have a feel for what is normal in terms of frequency, severity and type of infection in childhood. With the changing demographics of childcare moving towards larger centres, the average infant aged <1 year can be expected to have about 10 respiratory infections per year and a significant proportion have an associated otitis media.6 However, if the child is generally growing and developing appropriately, and the infections settle without the need for complicated antibiotic regimens,6 this is unlikely to suggest immunodeficiency.
Most severe infections occur as single events in otherwise healthy children and are unlikely to relate to compromised host defences; however, more than one serious or life-threatening infection, a single serious infection with an unusual or low pathogenicity organism (eg Pneumocystis jirovecii pneumonia), or an unusual site of infection (eg liver abscess) are important potential indicators of an inadequate immune system. A thorough history is therefore needed to elucidate the number and severity of infections. It is also important to consider differential diagnoses of recurrent infections, such as defects that block drainage of secretions. The causes may be anatomical (eg nasal defects causing recurrent sinusitis), extrinsic (eg inhaled foreign objects) or physiological (eg failure of the mucociliary escalator, causing recurrent chest infections in cystic fibrosis). The clue here is that blockages usually affect the blocked site only, as opposed to immune defects where multiple sites may be involved.
The type of infection may hint at the underlying mechanism of PID. Disorders involving B cells result in antibody deficiency, with sinopulmonary infections caused by encapsulated organisms such as Streptococcus pneumonia or Haemophilus influenzae. If severe, these infections will present at 3–6 months of age, as maternal antibody passively acquired across the placenta begins to wane (eg X-linked agammaglobulinaemia). If less severe, they present later (eg common variable immunodeficiency [CVID]).
T-cell or combined immunodeficiency disorders may also show a lack of antibody and bacterial infections, but added to this is an inability to fight viral and fungal infections (eg Varicella zoster virus (VZV), refractory candidiasis). Severe T-cell deficiency, such as severe combined immunodeficiency (SCID), may also present with failure to thrive or developmental delay.1
Disorders of phagocytic function result in recurrent soft-tissue infections caused by pyogenic bacteria and/or fungi at multiple sites,7 including the lungs, skin and lymph nodes, as well as refractory gingivitis. Complement deficiencies result in infections caused by encapsulated pyogenic bacteria, especially Neisseria species and should be considered in any child with recurrent meningococcal disease.
Finally, some PIDs occur as part of a syndrome. For instance, DiGeorge syndrome, caused by thymic aplasia, also results in facial and cardiac abnormalities, and hypoparathyroidism, whereas Wiskott-Aldrich syndrome manifests with the triad of immunodeficiency, eczema and thrombocytopaenia.
For SIDs, a detailed exploration of the risk factors and review of the patient’s history is warranted to identify possible causes of this. HIV should be considered in children presenting with infections where the mother has been inadequately screened in pregnancy and, in particular, in women arriving from HIV-endemic countries.
Examination
Examination is largely unremarkable in milder, isolated immunodeficiencies without syndromic features. Children may fail to thrive or be underweight in severe cases. Examination of the skin may show abscesses or persistent viral infections (eg warts), while eczema or eczema-like rashes occurs in a number of PIDs.8 There may be hepatosplenomegaly and lymphadenopathy associated with some conditions (eg CVID, HIV). Reduced or absent tonsils and lymph nodes occur in severe B-cell or combined immunodeficiency disorders, although normal infants may often have difficult-to-palpate lymph nodes. Finally, chronic infection results in chronic organ damage; the most important clinical findings are deafness associated with recurrent ear infection and bronchiectatic lung changes.
Basic immunological investigations
A full blood count is a good start, potentially revealing lymphopaenia (seen in combined immunodeficiencies), neutropaenia (eg congenital or acquired neutropaenia) or thrombocytopaenia (eg Wiskott-Aldrich syndrome). Quantification of serum immunoglobulins (IgM, IgG, IgA and IgE) is cheap and potentially informative, but it is important to remember that antibody levels are generally lower in early life (Figure 1). Total IgG is generally the most important measurement, whereas the importance of an isolated low IgA is debatable and IgG subclasses are of uncertain significance. Commonly, a child may have relatively normal levels of antibody but still be unable to mount an appropriate antibody response to infection. This condition is called specific antibody deficiency. In this case, specific antibody levels are measured pre- and post-vaccination with well-defined vaccines (eg diphtheria, Pneumococcus).
Immunophenotyping of lymphocyte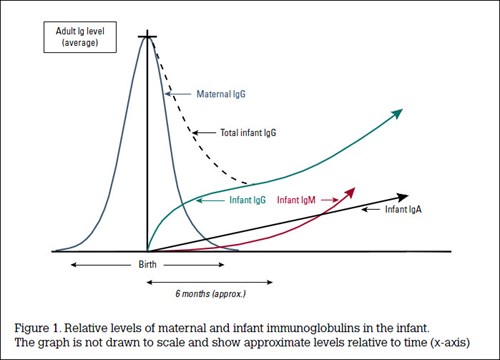 subpopulations using flow cytometry can help to define both the presence and type of immunodeficiency, in particular cellular immunodeficiency. If a T-cell deficiency is suspected, then T-cell stimulation assays may be performed. Finally, there is an array of tests that allow assessment of neutrophil function (eg oxidative burst assay) in the assessment of chronic granulomatous disease.
subpopulations using flow cytometry can help to define both the presence and type of immunodeficiency, in particular cellular immunodeficiency. If a T-cell deficiency is suspected, then T-cell stimulation assays may be performed. Finally, there is an array of tests that allow assessment of neutrophil function (eg oxidative burst assay) in the assessment of chronic granulomatous disease.
Inherited deficiencies of the complement pathway cause sinopulmonary infections and, potentially, autoimmune disease, and are best tested by measuring the total classical and alternative complement pathway activity. Finally, as with much of paediatrics, where complex diseases often have a single genetic aetiology, the future of single-gene PID diagnosis is likely to be the increasing use of genetic screening, particularly with the rapid emergence of genome-based screening techniques, which may allow us to screen for hundreds of PID-associated genes concurrently.
With regard to SID, in the correct clinical and demographic context, the combination of CD4+ T-cell deficiency with hypergammaglobulinemia could suggest possible HIV/AIDS for which serological and molecular testing is readily available; however, this is beyond the scope of this review.
Conclusion: when to refer for investigation of PID?
This review is not exhaustive and when trying to gauge whether a child has an immunodeficiency, there are a number of useful clinical instruments available, the most widely used of which comes from the Jeffrey Modell Foundation9 (Table 1). Referral to a specialist immunologist for further assessment is warranted should the patient fit these criteria, particularly if screening tests support a PID and an alternative aetiology is not identified.
Table 1. The Jeffrey Modell Foundation 10 warning signs of possible childhood immunodeficiency9
|
|
1. ≥4 ear infections within 12 months
|
|
2. ≥2 serious sinus infections within 12 months
|
|
3. ≥2 pneumonias within 12 months
|
|
4. ≥2 deep-seated infections including septicaemia
|
|
5. Recurrent deep skin or organ abscesses
|
|
6. Persistent oral or skin fungal infections
|
|
7. Infection does not respond to ≥2 months of standard antibiotics
|
|
8. Intravenous antibiotics needed to clear infections
|
|
9. Failure to thrive or growth retardation
|
|
10. Family history of PID
|
Illustrative case
A Caucasian boy aged 10 months has had recurrent infections from age 4 months. These included a persistent mucopurulent nasal discharge, recurrent otitis media with discharge and cough associated with fever. The infections were responsive to antibiotics but with incomplete resolution of symptoms and recrudescence on withdrawal. On examination, he was non-dysmorphic, had a perforated left eardrum and was failing to thrive, falling from the 50th to 3rd centile for weight. The family history included the mother’s older brother dying in early infancy from infection.
Case analysis This history of recurrent mucopurulent sinopulmonary infections suggests antibody deficiency and the age of onset suggests a severe B-cell deficiency associated with the loss of transplacental maternal antibody (Figure 1). In a boy, this is most likely to be X-linked agammaglobulinemia, which is supported by the presence of an X-linked family history. This was indeed confirmed by the absence of IgG/IgA/IgM and B-cells and by sequencing of the BTK gene, which showed a mutation.
Key points
- Immunodeficiencies are classed as either primary or secondary; primary immunodeficiencies are particularly important for children.
- Although recurrent infections are a sign of possible immunodeficiency, they are common in normal children, and very young infants potentially have up to 10 respiratory infections a year.
- GPs should be aware of the clues in the history that suggest a possible immunodeficiency in order to guide their clinical work-up.
Competing interest: None.
Provenance and peer review: Not commissioned, externally peer reviewed.