Case
A Caucasian man, 67 years of age and with no relevant past medical history, presented with progressive oral pigmented lesions and pigmented fingernails, which had appeared several years ago. He was a non-smoker and he did not take any medication. None of his family members had the same lesions. He did not have any bowel diseases. Physical examination revealed some 5-mm brown macules on the hard palate (Figure 1), longitudinal hyperpigmented bands on almost all the fingernails, and dark brown pigmentation on the proximal nailfolds (Figure 2). Laboratory tests did not show any alterations.
Question 1
What is the most likely diagnosis?
Question 2
What are the main differential diagnoses?
Question 3
What is the most appropriate management?
Answer 1
The most likely diagnosis is Laugier-Hunziker syndrome (LHS). It is a rare disease of unknown incidence, characterised by diffuse, hyperpigmented macules of the oral mucosa, lips, hard palate, tongue and gingiva, and longitudinal melanonychia. However, nail pigmentation is also present in 50% of the cases and pigmentation can appear in other locations such as the neck, genitals, perineum, eyes, trunk and oesophagus.1 Pseudo-Hutchinson sign refers to the brown pigmentation of the proximal nailfold. This must be distinguished from the Hutchinson sign, which is seen in nail melanomas.2
LHS is a benign disease that has no systemic manifestations or potential for malignancy. It is seen in middle-aged adults and the rate of occurrence is the same in men and women. This condition has been reported to be sporadic but an inherited autosomal dominant pattern has also been described.3 Histology shows basal cell hypermelanosis, increased melanophages in the papillary dermis, pigment incontinence, melanocytes that are normal in number and morphology, although some reports have described an increase in the number of melanocytes. Naevus cells have not been reported.
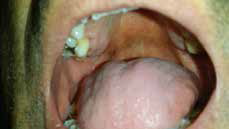 |
| Figure 1. Brown macules on the hard palate |
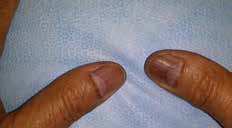 |
| Figure 2. Longitudinal hyperpigmented bands on the nails and dark brown pigmentation on the proximal nailfolds |
Answer 2
Differential diagnoses include:
- Peutz-Jeghers syndrome – The pigmentation is concentrated on the lips and oral mucosa, but unlike LHS, it appears by early childhood and does not involve the nails. Peutz-Jeghers syndrome follows an autosomal dominant inheritance pattern and is associated with benign bowel hamartomatous polyposis. There is an increased risk of developing malignancies, especially in the genitals, breast and lungs.4
- Drug ingestion – Drugs that can cause pigmentation include phenytoin, tetracyclines, amiodarone, oral contraceptives, ketoconazole, antimalarials, clofazimine, zidovudine, phenothiazine and some chemotherapeutics. Pigmentation usually occurs months-to-years after ingestion of the drug and tends to resolve once the drug is discontinued.5
- Albright disease – Pigmentation is located in the lips and genitals and is often unilateral. This syndrome also includes premature puberty and fibrous dysplasia.
- Primary adrenocortical insufficiency (Addison’s disease and adrenalectomy) – Pigmentation is related to sun-exposed areas of the skin, scars and mucosa resulting from elevated levels of adrenocorticotropic hormone (ACTH) precursors. Systemic symptoms, such as weakness, weight loss, asthenia, low blood pressure, hypoglycaemia and bowel changes, are also present.
Answer 3
LHS is a benign condition, so treatment is optional and can be used for cosmetic reasons. Treatments for the hyperpigmented macules include cryosurgery, Q-switched Nd:YAG and Q-switched alexandrite lasers. However, recurrence may occur after treatment. Sun avoidance is also recommended to prevent new pigmentations.6
Competing interests: None.
Provenance and peer review: Not commissioned, externally peer reviewed.