General practitioners are frequently faced with a broad range of differential diagnoses when a patient presents with chronic shortness of breath or chronic respiratory symptoms such as cough, sputum or wheeze. Before considering a diagnosis of lung disease, other diagnoses including heart failure and anaemia, and simple poor fitness, need to be considered. Even when the focus is more likely lung disease, there remain a number of conditions including chronic obstructive pulmonary disease (COPD), asthma, interstitial lung disease and pulmonary hypertension that need to be considered. Bronchiectasis should be included in this broad list of differentials.
While clinicians may automatically think of children and young adults with cystic fibrosis (CF) when bronchiectasis is mentioned, it is now recognised that there are an increasing number of patients who are diagnosed with non-CF bronchiectasis when they reach adulthood. Delay in the diagnosis, investigation and management of bronchiectasis is common and this delay has been shown to be associated with more rapid progression of disease.1,2
This article will focus on non-CF bronchiectasis and aims to provide advice regarding in whom to suspect bronchiectasis and how to proceed with confirming or refuting a diagnosis. It also discusses the principles of management to minimise disease progression and the management acute exacerbations, symptoms, and the associated disability and impaired quality of life.
However, before addressing these areas it is first important to outline the concepts that underlie the pathology, pathophysiology and diagnosis of bronchiectasis.
What is bronchiectasis?
Bronchiectasis is classically defined as a pathological diagnosis typically confirmed by radiology. Bronchiectasis is characterised by abnormal, irreversible bronchial dilatation or a fixed increase in airway diameter. Bronchiectasis is currently usually diagnosed by a chest high-resolution computed tomography (c-HRCT) scan (Figure 1).
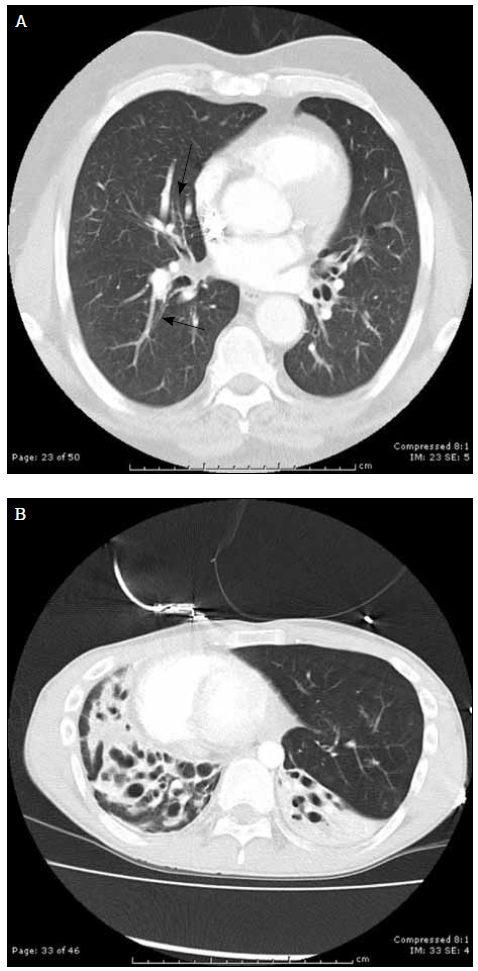
Figure 1. Changes of bronchiectasis on c-HRCT can be subtle. A) Dilatation and loss of normal tapering of right middle lobe bronchi (arrows) or obvious; B) Bilateral saccular dilatation of bronchi with associated collapse and parenchymal destruction
While the primary site of damage detected by c-HRCT is the larger airways, this is likely to be a later or parallel manifestation of a disease process involving other components of the lung, including the smaller airways not well visualised by c-HRCT and the bronchial mucosa. Particularly in the earlier stages of disease, chronic airway infection and inflammation consistent with bronchiectasis may not be accompanied by airway dilatation on c-HRCT, as seen in children who are at an elevated risk of developing later radiologically confirmed bronchiectasis. Chronic suppurative lung disease (CSLD) is an all-encompassing term, used particularly in children, to define a clinical syndrome of chronic airway inflammation and suppuration with or without evidence of bronchiectasis on c-HRCT.
Given that bronchiectasis is a pathologic diagnosis it is possible to have evidence of bronchiectasis on c-HRCT without symptoms of chronic airway inflammation and suppuration. This may be related to relatively quiescent or minor disease or the fact that the cause of the airway dilatation is not related to a process affecting the airway directly, but rather a process involving the lung parenchymal with secondary fibrosis and retraction of the structures supporting the airway. This entity, typically seen in interstitial lung diseases and termed, 'traction bronchiectasis', is not usually considered as bronchiectasis per se unless there is evidence of associated airway suppuration and its management it typically directed at the underlying interstitial lung disease.
Burden of disease
Bronchiectasis can be caused by a broad range of disparate and esoteric conditions and is often idiopathic in nature. Although increasingly recognised, there is a lack of data regarding the burden of non-CF bronchiectasis in Australia. Aboriginal people living in remote Australia are at particular risk. While a study3 of Central Australia Aboriginal children found a prevalence of 1470/100 000, the estimated prevalence for Australians overall remains unknown. In the United States, estimates range from 4.2/100 000 in the 18–34 years age group to 272/100 000 in those over 75 years of age.4
The impact on the healthcare system also appears to be increasing. In the US, hospitalisations for bronchiectasis have increased between 1993 and 2006 and now occur at an annual rate of 16.5/100 000.5
In light of the limited understanding of the prevalence of bronchiectasis in Australia, it is impossible to assess the impact this has on disability and premature mortality at a national level.
A national bronchiectasis register being developed by the Australian Lung Foundation should facilitate more accurate estimates of Australian disease burden.
Pathophysiology
While the underlying cause of bronchiectasis may vary (or is even often undefined or idiopathic) it is ultimately due to injury to the airways, which is typically associated with chronic and recurrent inflammation because of an abnormality of airway anatomy, immunity or function. Despite extensive investigation, more than 80% of patients with bronchiectasis will have no clearly identified cause for their disease.6 Nonetheless, it is important to consider secondary causes as these may be associated with reduced progression if managed, can be associated with other nonrespiratory complications, or can have a familial association. The range of possible causes of bronchiectasis is listed in Table 1.7
Table 1. Aetiologies and factors associated with bronchiectasis7
- Congenital causes (eg. Mounier-Kuhn syndrome, Young syndrome)
|
- Chronic obstructive pulmonary disease and smoking
|
|
|
- Mucociliary dysfunction (eg. primary ciliary dyskinesia)
|
- Primary or secondary immune deficiency (eg. hypogammaglobulinaemia, lung and bone marrow transplantation, malignancy, HIV/AIDS, HTLV1)
|
- Pulmonary fibrosis and pneumoconiosis (eg. silicosis)
|
- Postobstruction (eg. with a foreign body)
|
- Postinfection (eg. tuberculosis, adenovirus, recurrent pneumonia)
|
- Recurrent small volume aspiration (eg. from upper airway secretions or gastric contents)
|
- Allergic bronchopulmonary aspergillosis
|
- Systemic inflammatory diseases (eg. rheumatoid arthritis, sarcoidosis)
|
Overlap syndromes
Bronchiectasis can frequently occur in parallel with more common forms of chronic lung disease including COPD and asthma. The increasing availability and use of c-HRCT has shown that up to 50% of patients with severe COPD will have co-existent bronchiectasis.8 In an Australian cohort of adult bronchiectasis patients, 15% had a co-existent diagnosis of asthma or COPD and 25% demonstrated significant (>15%) improvement with bronchodilators, which is suggestive of airway hyperactivity if not asthma.6 Bronchiectasis is more likely to co-exist with COPD in patients with moderate to severe disease, an acute exacerbation requiring hospitalisation in the past year, and if sputum contains potential pathogens even when clinically stable.9 There nonetheless remains debate whether this is indeed 'true' bronchiectasis or coincidental airway dilatation associated with advancing age or associated hypoxaemia.10,11
When to suspect bronchiectasis
Given the broad range of conditions that can cause bronchiectasis, the fact many patients can have idiopathic disease, the presence of mild disease and overlap syndromes, it is not surprising that it is difficult to identify which patients warrant further investigation.
The first group in whom to suspect bronchiectasis are those with a confirmed diagnosis of a condition that may predispose to disease, such as a confirmed immune deficiency. Another group that may not immediately spring to mind is men with primary infertility, particularly when related to azospermia or immotile sperm. In these cases, milder forms of CF, Young syndrome or primary ciliary dyskinesia/immotile cilia syndrome should be considered.
More difficult and more common is the patient presenting with chronic respiratory symptoms, especially cough and sputum production. While there is no absolutely accurate way to differentiate between chronic bronchitis, COPD and asthma and bronchiectasis (and indeed as noted all these conditions can co-exist with bronchiectasis) a number of clinical features may help and these are listed in Table 2.
- Diagnosis of asthma that is unresponsive to usual management
Table 2. Features that may suggest bronchiectasis in a patient presenting with chronic respiratory symptoms
- Digital clubbing (this is rare in COPD and asthma)
|
- Lack of a significant smoking history (less than an average of 20 cigarettes per day for 10 years) in a person with suspected COPD
|
- History of recurrent and/or severe pneumonia including tuberculosis
|
- Presence of 'unusual organisms' in sputum (eg. Aspergillus, atypical/nontuberculous mycobacteria, Pseudomonas aeruginosa, Escherichia coli, Klebsiella pneumoniae)
|
- Childhood associated with significant environmental and social disadvantage*
|
| * This includes Aboriginal and Torres Strait Islander people, as well as people who have immigrated from low income countries.52 In this group of people, tuberculosis as the cause of chronic respiratory symptoms should also be considered |
Case study continued
You examine Jane and she has bibasal crackles, occasional wheeze, an oxygen saturation of 95% and no digital clubbing. You perform spirometry. It is of good quality and reproducibility and demonstrates moderate airflow obstruction that is not fully reversible with salbutamol. A plain chest X-ray is reported as demonstrating minor right basal fibrotic changes. Given the presence of wheeze and airflow obstruction you suspect a combination of asthma with an element of COPD. You commence Jane on a short acting beta-agonist and combination long acting beta-agonist/inhaled corticosteroid inhaler. On review 1 month later her condition has not improved.
Diagnosis and assessment
In general, a diagnosis of bronchiectasis requires a clinically consistent history in association with evidence of fixed and abnormal bronchial dilatation. As highlighted earlier, evidence of bronchial dilatation is typically confirmed on c-HRCT (Figure 1).
Plain chest X-ray
While plain chest X-ray may demonstrate large airway dilatation it is not sensitive (ie. a normal chest X-ray does not exclude bronchiectasis) nor does it provide an accurate measure of the extent of disease or any associated interstitial damage (eg. pulmonary fibrosis).
Chest high-resolution CT scan
Chest HRCT allows an assessment of a number of features of airway anatomy including diameter, wall thickness and the presence or absence of normal tapering. Despite its benefits, c-HRCT has inherent limitations, such as the inability to confirm fixed airway dilatation on a single study. In patients with concurrent infection, c-HRCT may demonstrate temporary airway dilatation that may resolve on subsequent studies. To avoid the need for repeated c-HRCT it is recommended diagnostic c-HRCT be performed when a patient is clinically stable and not in the setting of an acute exacerbation.
The definition of what is an abnormally dilated airway can provide further room for confusion. In general, this is said to be present in adults when the internal diameter of the airway on c-HRCT is greater than that of the accompanying branch of the pulmonary artery (termed a 'bronchoarterial ratio greater than one').12,13 While this is an important and accepted definition of airway dilatation used in reporting c-HRCT, it is also apparent this ratio increases with age11 and other factors including altitude and co-existent hypoxia.10 Too strict adherence to this ratio may produce poorer diagnostic sensitivity for c-HRCT in younger people and lower specificity in older adults. In general, if the results of c-HRCT are discordant with expectations or the changes are incidentally noted in a patient without clinical features of bronchiectasis, then further discussion with the reporting radiologist and a respiratory physician is warranted.
While the adage 'manage the patient not the X-ray' should be applied to patients without symptoms and only minor and incidentally detected changes of bronchiectasis on c-HRCT, it should be noted that bronchiectasis is often a progressive condition where minor changes can potentially be a harbinger of the development of later more extensive disease. Careful clinical review, searching for secondary causes if any chronic respiratory symptoms are present, and ongoing clinical follow up is warranted. In otherwise well patients with normal spirometry, a case may be made for follow up c-HRCT after 2 years to confirm a lack of radiologic progression.
The final issue that should be considered before ordering a c-HRCT is radiation. A c-HRCT has an effective radiation dose of up to 8 mSv, the equivalent of 400 plain chest X-rays or 3.6 years of background radiation.14,15 This is of particular importance for children and younger adults where the effective radiation dose and the lifetime implications associated with cancer induction are higher. Before ordering a c-HRCT in children and younger adults, specialist input is encouraged to ensure the potential risk of the radiation exposure is balanced by any possible benefit.16
Spirometry
Unlike conditions such as COPD, where the demonstration of airflow obstruction is an essential component of the diagnosis, in bronchiectasis, spirometry findings can vary and even be normal in the early stages of disease. Bronchiectasis is not always associated with evidence of airflow obstruction – as demonstrated by a reduced ratio of the volume expired in the first second (FEV1) compared with the total expired volume (FVC) during a forced expiration from total lung capacity on spirometry. A restrictive (reduction in both FEV1 and FVC with normal or elevated FEV1/FVC ratio) or mixed obstructive/restrictive pattern may often seen. While spirometry and more detailed lung function testing is useful in assessing severity of disease, monitoring progression and predicting prognosis (particularly in adults) it cannot be used to exclude or confirm a diagnosis of bronchiectasis.
Although there is no agreed definition for stratifying the severity of bronchiectasis on spirometry alone, it is reasonable to base this, in part, on the severity of impairment in FEV1 used in the Australian COPD guidelines17 as outlined in Table 3.
Table 3. Spirometry based stratification of bronchiectasis severity based on the COPDX Plan17
| Severity |
% predicted FEV1* |
| Mild |
60–80% |
| Moderate |
40–59% |
| Severe |
<40% |
| * FEV1 expiratory volume in the first second of a forced expiratory manoeuver |
Investigating for secondary causes
The broad range of secondary causes of bronchiectasis has been outlined in Table 1.7 The minimum recommended investigations for these is listed in Table 4.7 While all patients with a confirmed diagnosis of bronchiectasis warrant exclusion of secondary and preventable causes, many of these investigations require specialist support. The investigations that might be performed before specialist referral to expedite management are highlighted in bold.
Table 4. Recommended investigations for secondary causes of bronchiectasis7
|
|
- Immunoglobulin classes IgG, IgA, IgM, and IgG subclasses
|
- Sputum culture including mycobacterial culture
|
- Serological tests for Aspergillus and total IgE level in adults, especially if there is a history of wheeze/asthma
|
- Test for primary ciliary dyskinesia in children
|
| In addition, consider the following: |
|
|
- Test for cystic fibrosis transmembrane conductance regulator gene mutations
|
- Bronchoscopy for foreign body or airway abnormality and to obtain specimens for culture of respiratory pathogens, including mycobacteria
|
|
|
- Additional immunological tests – total IgE level in children, neutrophil function tests and lymphocyte subsets, and antibody responses to protein and polysaccharide antigens
|
- Test for primary ciliary dyskinesia in adults
|
|
|
| Note: suggested investigations before specialist referral are highlighted in bold |
Case study continued
Given Jane's condition has not improved you call your local respiratory physician. She suggests full blood examination (FBE), immunoglobulin levels, aspergillus serology and total IgE, one sputum for usual microbiology and three sputums for atypical mycobacterial microbiology, and a c-HRCT. She arranges an appointment to see Jane in 6 weeks with the results.
Management of bronchiectasis in primary care
The complexities associated with suspecting and confirming a diagnosis of bronchiectasis and assessing for secondary causes have already been highlighted. Even once this is achieved, the ongoing monitoring and management of bronchiectasis includes a plethora of treatment options. This section outlines available therapies and provides advice as to when they should be considered.
The principles of ongoing management of bronchiectasis are based on the monitoring of severity, reducing progression and complications, early treatment of acute exacerbations, minimising disability, considering transplantation in appropriate patients, managing comorbidities and early utilisation of palliative care services when necessary. As with any complex chronic disease, patients benefit from a multidisciplinary care approach. In the case of bronchiectasis, this may comprise input from a respiratory physician, a physiotherapist, a palliative care and mental health team and respiratory nurse.
When to refer
In general, all patients should be discussed or referred to a specialist paediatric or adult respiratory physician for initial assessment and advice regarding the development of an individualised management plan. In the case of children and young adults this should occur when the diagnosis is suspected and before organising a c-HRCT. While a specialist review may be difficult for some patients, a focused telephone discussion or telehealth consultation can often suffice.
The role of antibiotics
While bacteria and fungi are often found in the sputum of people with bronchiectasis, their role in disease development, acute exacerbations and progression is variable. In some cases they may be contributors to airway inflammation, damage and the progression of bronchiectasis (the 'vicious cycle' hypothesis).18 However, such organisms may also be commensals, colonising damaged airways but not contributing to acute exacerbations or progressive airway damage. Decisions regarding the use of antimicrobials must therefore be based on an individual patient's response in addition to the results of airway microbiology.
Acute exacerbations
While there is little evidence to support the use of antibiotics in acute exacerbations of bronchiectasis, there is general consensus that they should be used.19 An acute exacerbation can be defined as two or more of: increasing cough, shortness of breath, increasing volume/purulence of sputum. An unexplained significant (>10%) reduction in FEV1 or FVC over days or weeks should also raise suspicion of an acute exacerbation. In a patient who is not unwell or at risk of sudden deterioration (Table 5), initial oral antibiotic therapy is reasonable. The choice of antibiotic should be based on the most recent sputum culture. If this result is negative or not available, commencing treatment with amoxicillin-clavulanate or doxycycline is recommended.7 The course of therapy should be prolonged (at least 10 days). Early follow up (within 4 days) and regular review is required to ensure response and to consider inpatient management early if there is deterioration. While the rate of response varies, most patients would be expected to begin to improve within 7 days, although it can take up to 4 weeks to return to a baseline state.
Table 5. Features of an acute exacerbation of bronchiectasis that should prompt early discussion regarding referral for inpatient management
|
|
- Hypotension (systolic BP <90 mmHg or diastolic BP
<60 mmHg)
|
- Respiratory rate ≥30/minute
|
- Previous need for noninvasive ventilation/ICU
|
- Failure to improve after 7 days of oral therapy
|
- Hypoxia (new onset of oxygen saturation ≤93% on room air)
|
|
|
- Severe disease (FEV1 <40% predicted)
|
- Limited home and social supports or difficulty ensuring follow up/review
|
- Substantial disability (new onset of being unable to meet self care needs)
|
Long term and other antibiotic dosing strategies
There remains debate regarding the role of long term suppressive, intensive intermittent or eradicative antibiotic therapy in non-CF bronchiectasis and whether these can reduce acute exacerbation frequency and or severity, or disease progression, or enhance quality of life or survival. While a systematic review suggested little overall benefit20 from long term suppressive antibiotics, more recent studies have suggested macrolides, including azithromycin, can reduce acute exacerbation frequency and improve lung function.21,22 In patients with frequent exacerbations (three or more per year), a trial of long term antibiotic therapy may be considered. However, the use of antibiotics in this way is outside approved indications, can pose significant cost to patients and is best undertaken in partnership with a specialist respiratory service, which can organise hospital based supply of azithromycin with achievement of clear clinical endpoints before a decision regarding ongoing use. Any potential benefits need to be balanced against the risks associated with antibiotic side effects, drug interactions and the development of microbial resistance.
An alternative approach, particularly utilised in CF related bronchiectasis, has been the regular and intermittent use of intensive antibiotic therapy (typically intravenous) in stable patients.23 Such regular 'tune ups', while commonly used in CF, are supported by little evidence and not routinely recommended for non-CF bronchiectasis.
An additional strategy is intensive and prolonged antibiotic treatment with the aim of eradicating Pseudomonas aeruginosa when it is first isolated. This is based on the premise that P. aeruginosa is associated with more difficult and expensive treatment of acute exacerbations in the long term and worse outcome both in CF and non-CF related bronchiectasis. There is evidence in CF that prolonged nebulised, oral and/or intravenous antibiotics can lead to at least short term eradication of both nonmucoid24–27 and mucoid28 strains of P. aeruginosa. In turn, this is associated with a slower decline in lung function.28 A similar strategy of early and aggressive treatment of
P. aeruginosa outside the setting of an acute exacerbation in non-CF bronchiectasis has not been shown to be effective. Nonetheless, despite the lack of evidence, some specialist respiratory bodies recommend attempting eradication.29 In light of this controversy, it is reasonable to seek specialist advice regarding eradicative strategies when P. aeruginosa is initially isolated in patients with non-CF bronchiectasis. Regimens can involve a combination of longer term oral and nebulised antibiotics and can be provided in the community under the supervision of the primary care doctor.
Atypical mycobacterial disease
Mycobacteria other than M. tuberculosis and M. leprae (termed atypical mycobacteria or nontuberculous mycobacteria [NTM]) are generally free living environmental organisms that, unlike
M. tuberculosis, are not associated with person-to-person transmission. The NTM most commonly associated with lung disease in patients without HIV/AIDS is M. avium complex (MAC) with
M. kansasii and M. abscessus being less frequently encountered.30
It is unclear whether other NTM can cause lung disease, in particular bronchiectasis in patients without HIV/AIDS.
While the exact contribution of NTM in the development or progression of bronchiectasis is poorly understood, these organisms (particularly MAC) are increasingly noted in the sputum of people with bronchiectasis and can be associated with progressive lung damage. While NTMs can be isolated in the sputum of people with pre-existing lung disease, they can also be associated with the development of progressive lung disease, typically in older women, without pre-existing disease. This syndrome is characterised by pulmonary infiltrates, nodules and bronchiectasis with a predilection for the lingular segment of the left upper lobe and the right middle lobe.31
Also termed 'Lady Windermere syndrome' in reference to Oscar Wilde's play Lady Windermere's Fan it is hypothesised that this is caused by suppression of cough in self conscious, overly polite women.32
Differentiating airway colonisation with NTM from active disease requiring treatment is difficult and requires specialist input. It is based on consistent or significant isolation of the organism with typical and progressive radiologic changes.30 If NTM are thought to be driving the development or progression of bronchiectasis, treatment may be considered. Nonetheless, it is prolonged, expensive and complicated, typically involving three-drug therapy for 1–2 years and until the sputum remains negative for NTM for 1 year. Macrolide therapy is an important component of most treatment regimens and the use of prior long term suppressive macrolide therapy (such as azithromycin) may result in the selection of macrolide resistant NTMs with associated less effective and more prolonged and complicated therapy.
Inhalational and other therapies
Inhalational therapy for bronchiectasis can be divided into agents typically used in asthma and COPD, inhalational antibiotics and those used to enhance sputum clearance. Given the existence of overlap syndromes it is not surprising that many patients may benefit from management relating to asthma or COPD. If there is felt to be co-existant asthma (demonstrated by wheeze and a significant >12% and 200 mL increase in FEV1 with bronchodilators) or COPD (emphysema seen on c-HRCT or significant smoking history) then a trial of short and long acting beta agonists, anticholinergics and/or inhaled corticosteroids with assessment of response many be warranted. While there is some evidence that inhaled corticosteroids, with or without associated long acting beta-agonists, may reduce symptoms in non-CF bronchiectasis, this effect is small. Overall there is little evidence to support the routine use of these inhalational therapies, particularly inhaled corticosteroids, in patients without co-existent asthma or COPD.33,34
Inhalational antibiotics have often been used in bronchiectasis. Long term use of nebulised gentamicin for 12 months in patients with non-CF bronchiectasis who have chronic sputum bacterial colonisation and at least two acute exacerbations per year has been shown to reduce sputum bacterial colonisation (which is associated with poorer outcome), improve exercise tolerance and reduce acute exacerbation frequency.35 While a trial of inhaled gentamicin in such patients may be warranted, this should be commenced in consultation with a specialist respiratory service given this is not a licensed use for gentamicin in Australia and may result in the development of initial worsening of lung function in some patients. Inhalational antibiotics are also often an adjunct to oral antibiotics or home based intravenous antibiotics in the primary healthcare based management of acute exacerbations of bronchiectasis and as eradicative or suppressive therapy in selected patients.
Mucolytics and other agents used to enhance sputum clearance that have been considered for non-CF bronchiectasis include recombinant human deoxyribonuclease, bromhexine, n-acetyl cysteine and hyperosmolar agents including hypertonic (6–7%) saline and mannitol. In contrast to CF related bronchiectasis, recombinant human deoxyribonuclease has not shown benefit in non-CF bronchiectasis and indeed may be harmful with greater acute exacerbation frequency and decline in lung function.36 This highlights the fact that interventions shown to be effective in CF related bronchiectasis cannot necessarily be extrapolated to non-CF bronchiectasis.
While mannitol37 and hypertonic saline have shown promise in CF related bronchiectasis, their role in non-CF bronchiectasis is yet to be well demonstrated.38 Hypertonic saline demonstrated short term benefits in non-CF bronchiectasis39 but was not superior to isotonic saline based on quality of life, lung function and acute exacerbation frequency in long term studies.40 Pending the outcome of current trials the use of mucolytics and other agents used to enhance sputum clearance in non-CF bronchiectasis should be limited to those patients in whom sputum expectoration is a major issue and occur in consultation with a specialist respiratory service.
While there is little evidence specifically relating to bronchiectasis and the benefit of pneumococcal and influenza vaccination, the risk of concomitant infection in patients with pre-existing lung disease would support their use in all patients. Children and adults with bronchiectasis should have enhanced influenza and pneumococcal vaccination schedules.41
Physiotherapy and pulmonary rehabilitation
Physiotherapy to enhance sputum clearance has been a longstanding mainstay of management and there is evidence it improves cough, exercise tolerance and, in children, lung function.42,43 Given the risk of silent aspiration, head-down sputum clearance techniques are now discouraged.
There is evidence that pulmonary rehabilitation and tailored exercise programs improve exercise tolerance in people with bronchiectasis.44 Patients with bronchiectasis affecting their exercise tolerance or activities of daily living should be referred for pulmonary rehabilitation and/or have a tailored exercise program developed in consultation with a physiotherapist. This may occur in the community using available local exercise facilities.
Other forms of physiotherapy-based intervention including focused inspiratory muscle training have not shown benefit in bronchiectasis and are not advocated.44,45
Severe disease
Many people with bronchiectasis will have progressive disease1,6 and develop severe bronchiectasis. While all the aspects of management outlined have a clear role in those with severe disease, a range of additional interventions may be appropriate. In turn, it is important that people with bronchiectasis are regularly reviewed to assess the severity of their disease. Such assessment should, as a minimum, include assessment of disability, frequency of acute exacerbations and spirometry. While there is no agreed spirometry based classification for bronchiectasis severity, an FEV1 <40% should prompt enhanced monitoring for complications of severe disease and, if it has not already occurred, discussion with a specialist respiratory service.
Domiciliary long term oxygen therapy
In patients with significant disability or a severe reduction in FEV1 (<40% predicted) regular clinical review should include an assessment of suitability for domiciliary oxygen therapy. While there is no evidence specifically relating to the benefits of long term oxygen therapy in bronchiectasis, this can reasonably be extrapolated from the evidence available for COPD46 and supported by Australian recommendations.47 While assessment for long term oxygen therapy usually requires arterial blood gas analysis, in general, if the oxygen saturation of is >93% on room air when the patient is clinically stable then an arterial puncture is unlikely to reveal sufficient hypoxaemia to warrant this therapy. If the oxygen saturation is 93% or less in nonsmoking patients, then referral to a specialist respiratory service for assessment of the appropriateness of long term oxygen therapy should occur.
Surgery and transplantation
In the past, localised resection/lobectomy of focal bronchiectasis was occasionally undertaken in the hope this would reduce symptoms and prevent extension of disease to unaffected lobes. Local resection remains an option for the management of localised disease and is is generally reserved for bronchiectasis related to NTM resistant to medical management.48 Nonetheless, 26% of these highly selected patients will develop recurrent and progressive disease following surgery.49 If considered, such surgery should be undertaken by specialist respiratory and cardiothoracic services with experience in this area.
In selected patients with severe bronchiectasis lung transplantation also has a role. While there are no national criteria regarding suitability for transplantation, patients aged less than 65 years with severe disease and few or no comorbidities should be assessed for suitability. While many patients with non-CF bronchiectasis may not be suitable for transplantation, in those who are transplanted, the peri-operative mortality is low (<2%) and median survival is greater than 5 years.50
Palliative and end-of-life care
Palliative care focusing on symptomatic management, particularly relating to shortness of breath, anxiety and sputum production, can be invaluable in improving quality of life. In patients with severe disease with symptoms resistant to the medical management as outlined above, fans, benzodiazepines, narcotics and anticholingerics can reduce symptoms. As with any chronic disease, bronchiectasis is often complicated by depression and early detection and treatment of this can also enhance quality of life.
In patients with advanced disease in whom transplantation is not an option, the development of a health management directive with the patient and family is important to ensure acute exacerbations are appropriately and sensibly managed. Management of advanced disease is also often aided by involvement of a palliative care team to facilitate end-of-life care, aid symptom management and provide additional home based support. Given the heterogeneous nature of bronchiectasis and its gradual progression, it is extremely difficult to provide clear guidance regarding prognosis and patient survival. While this may make planning end-of-life care support difficult, there is a clear rationale for early referral to palliative care services for symptom control in patients with severe disease.
Prognosis
It is not surprising that in such a heterogenous group prognosis can vary greatly. Nonetheless, approximately 10% of adults with non-CF bronchiectasis will die within 5–8 years of diagnosis with this being directly attributed to their lung disease in over half.6,51 Factors associated with poorer prognosis include tobacco smoking, Gram negative organisms (especially Escherichia coli and P. aeruginosa) and aspergillus on sputum culture and greater impairment in FEV1 and FVC.34,51
Summary
While many patients with bronchiectasis present with chronic respiratory symptoms, there are a range of clinical factors that should prompt primary care clinicians to suspect this condition. A lack of a smoking history in someone with chronic cough and sputum production, 'asthma' resistant to therapy and unusual organisms on sputum culture should raise the suspicion of bronchiectasis. While c-HRCT is increasingly accessible it can pose its own problems. Minor disease in particular needs to be assessed in the clinical context and all scans are associated with a significant radiation dose that is of particular concern in children and younger adults. Once a diagnosis is confirmed it is important to evaluate for secondary causes. While many people will have no cause identified, in the minority who do, this can alter management and outcome. Given its complexity specialist advice is often useful.
The management of bronchiectasis is complicated, multifaceted and requires the coordination of a broad range of healthcare providers. As such it is ideally managed and coordinated in primary care with primary healthcare providers acting as brokers and coordinators of care. Management plans need to be tailored to the individual based on the severity of their disease, acute exacerbation frequency, sputum microbiology, respiratory and nonrespiratory comorbidities and patient preference. Given bronchiectasis is a chronic disease that can be associated with both long term disability, as well as less marked premature mortality, the importance of structured care as part of regular primary care review and reassessment is the key to successful management.
Key points
Bronchiectasis can present for the first time at any age and can often coexist with other lung diseases including COPD and asthma.
It is important to exclude and, where possible, treat secondary causes.
Diagnosis, assessment and ongoing management are best delivered as part of a multidisciplinary approach that includes primary, allied health and specialist respiratory care.
Ongoing management may include:
- management of acute exacerbations
- pneumococcal and influenza vaccination
- treatment of nontuberculous mycobacteria
- long term oral and inhaled antibiotics
- physiotherapy/exercise training/pulmonary rehabilitation
- palliative care
- surgery and transplantation.
Conflict of interest: none declared.