The disease has a peculiar geographical distribution, with the highest reported prevalence in northwest England, and is also common in Australia, New Zealand and North America, reflecting the high frequency of British ancestry in these countries.4 The disease is uncommon in other regions such as Scandinavia, Africa and Asia. Studies from the United Kingdom and New Zealand suggest that the prevalence and the severity of new cases have greatly decreased in the past 25–30 years.5,6 The reasons for this are unclear, but may reflect altered environmental factors or an increase in immigration from regions such as Asia, where PDB is rare. There are no recent prevalence studies of Paget disease in Australia.
Histologically, PDB is characterised by a marked increase in activity of both osteoclasts and osteoblasts. Bone remodelling is increased and disorganised, with persistence of woven bone at pagetic sites, resulting in bone which is structurally impaired and prone to deformity and fracture.
The aetiology of PDB is not well understood, but involves genetic and environmental factors. A family history of the disease is common in affected patients, and it can be inherited as an autosomal dominant trait.7,8 Mutations in the sequestome-1 (SQSTM1) gene have been identified in about one-quarter of kindreds with familial PDB, and in about 5% of patients with no family history of the disease.9 The SQSTM1 gene encodes a scaffolding protein, which plays an important part in development and activation of osteoclasts, but exactly how SQSTM1 mutations cause the characteristic focal lesions of PDB is unknown. Recent genome-wide association scan studies have identified a further seven susceptibility genes for PDB.10,11
For many years, infection with measles or other paramyxoviruses has been postulated as an environmental factor in the pathogenesis of PDB, but this remains controversial, with conflicting results from different laboratories.12 A recent study provided evidence, both from patients with PDB and a mouse model, that measles infection may interact with SQSTM1 mutations to produce the characteristic pagetic phenotype.13 Other possible environmental factors include local trauma to bone14 and environmental toxins.15
Diagnosis
Clinical features
The classic presentation of Paget disease (as described by Sir James Paget in 1877) as a painful, deforming disease is now uncommon. Nowadays, PDB is detected most frequently as an incidental finding on radiographs or on biochemistry (as raised alkaline phosphatase activity). Most commonly, the disease involves the axial skeleton, particularly the pelvis, lumbosacral spine, skull, femur or tibia, but any bone may be affected, and the disease may be localised to one or a few bones or widespread. The clinical presentation of PDB is summarised in Table 1 and appropriate investigations are shown in Table 2.
| Table 1. Clinical presentation of Paget disease of bone |
- Incidental finding on radiology or biochemistry
- Bone pain
- Arthopathy
- Deformity
- Fracture
- Deafness
- Neurological complications
- Osteosarcoma
|
Pagetic bone pain occurs in a minority of patients. In some cases this is constant, present at rest and poorly localised, with a dull, boring character, whereas other patients have localised, mechanical pain, worse on weight bearing, arising from microfractures or localised lytic lesions. Paget disease of the bone adjacent to a joint can cause a secondary osteoarthritis. Deformity, such as bowing of the femur or tibia, may be asymptomatic or associated with mechanical pain in the affected limb or the contralateral side, arising from secondary gait problems. Neurological complications are uncommon, but include deafness, which may be conductive, sensorineural or mixed. Malignant transformation of pagetic bone to osteosarcoma is rare, with a lifetime risk of less than 1%.
| Table 2. Investigation of Paget disease of bone |
- Plasma alkaline phosphatase
- Liver function tests
- Vitamin D levels
- Isotope bone scan
- Radiography of affected bones
|
Diagnostic imaging
Paget disease of the bone is diagnosed radiologically. Early in the course of the disease, lytic activity predominates, causing focal osteolytic lesions (osteoporosis circumscripta) or flame shaped, advancing lytic wedges in the long bones (Figure 1). Subsequently, areas of sclerosis develop, leading to the characteristic appearances of mixed lytic and sclerotic areas, thickened trabeculae, bone expansion, cortical thickening and deformity. Isotope bone scanning is more sensitive than plain radiology in detecting pagetic lesions, and is indicated in most newly diagnosed patients to determine the distribution of the disease. This allows the identification of involved bones with the potential for complications, such as the base of the skull, spine and long bones. Plain radiographs of abnormal bones as identified on the bone scan should then be performed to confirm the diagnosis and to assess severity and risk of local complications. The radiological appearances are usually characteristic, but occasionally a differential diagnosis of sclerotic or lytic metastases needs to be considered. In these cases, computerised tomography (CT) or magnetic resonance imaging (MRI) is generally diagnostic; bone biopsy is rarely required.
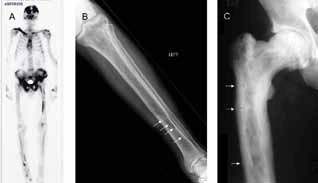
Figure 1. Radiological features of Paget disease of bone
Biochemistry
Plasma total alkaline phosphatase activity (ALP) is the most clinically useful indicator of disease activity in PDB. It is elevated in most untreated patients,16 but may be within the reference range in patients with monostotic or limited disease. Increased ALP activity is also seen in liver disease and occasionally from other sources, and in such cases ALP isoenzymes and bone resorption markers such as urine N-telopeptide are helpful in assessing pagetic activity. Serum 25-hydroxyvitamin D should be measured for two reasons: because osteomalacia may also present with bone pain and a raised alkaline phosphatase level, and because vitamin D deficiency should be corrected before bisphosphonate therapy to avoid the risk of hypocalcaemia.
Treatment
Simple analgesics and nonsteroidal anti-inflammatory drugs are helpful in managing symptomatic PDB, but the mainstay of treatment is bisphosphonate therapy. Bisphosphonates have been demonstrated to reduce bone turnover, improve bone pain, promote healing of osteolytic lesions and improve quality of life in patients with PDB.17,18 Whether bisphosphonate treatment prevents long term complications of the disease is unknown.
Not all patients with PDB require bisphosphonate treatment. Those with symptomatic disease should be treated, as there is good evidence that this responds to bisphosphonate therapy,19 and this is subsidised by the Australian Pharmaceutical Benefits Scheme (PBS). In some patients, it can be difficult to determine whether pain is arising from PDB or from coexisting conditions such as osteoarthritis. In such cases, a trial of therapy is reasonable if the pain localises to an area of pagetic involvement. A strong case can also be made for the treatment of asymptomatic patients in whom the location of disease (such as long bones, vertebrae or base of skull) puts them at risk of future complications. In addition, it may be reasonable to treat asymptomatic younger patients with involvement of articular surfaces (such as the hip joint) with the aim of preventing secondary arthritis. Pre-operative treatment with bisphosphonates is recommended for patients undergoing elective surgery (such as joint replacement) to bones with metabolically active Paget disease to reduce perioperative bleeding.20 Indications for treatment and recommended drug therapies are listed in Table 3.
| Table 3. Indications for treatment of Paget disease of bone and recommended drug therapies |
| Indications |
Pain in pagetic bones
Neurological complications
Significant osteolytic lesions
Involvement of long bones, vertebrae or base of skull
Before surgery involving pagetic bones
Significant joint involvement (eg. hip joint) |
| Recommended drugs |
Zoledronic acid: 5 mg single dose, given IV over at least 15 minutes
Alendronate: 40 mg daily, given orally for 3–6 months
Risedronate: 30 mg daily, given orally for 2 months |
The treatments of choice for PDB are oral alendronate, oral risedronate and intravenous zoledronic acid.21 Intravenous pamidronate has been largely superceded by zoledronic acid, and other drugs such as etidronate, tiludronate and calcitonin are largely obsolete.
A key clinical trial demonstrated superiority of a single infusion of intravenous zoledronic acid over a 2 month course of oral risedronate22–24 with normalisation of ALP in 89% and 58% of patients respectively, and greater improvements in pain and quality of life in the zoledronic acid group. Patients in this trial who achieved remission have been followed up for 6.5 years; relapse has occurred in 0.7% of patients treated with zoledronic acid compared with 20% for risedronate.22–24 Intravenous zoledronic acid is therefore probably the treatment of choice, although risedronate and alendronate remain good options for patients who prefer oral therapy, or if facilities for intravenous treatment are not available. When treating PDB, oral bisphosphonates are given at high dose for several months and stopped when remission is achieved; treatment should not be continued long term.
A common side effect of intravenous bisphosphonate treatment is a transient flu-like illness, occurring in one in 4 patients. Serious side effects are rare, but include acute renal failure and uveitis, and a history of uveitis is a contraindication. Oral bisphosphonates are generally well tolerated, but must be taken on an empty stomach, half an hour before food, and separately from other medications. They can cause dyspepsia and occasionally oesophageal ulceration.
The response to treatment should be monitored by measuring plasma alkaline phosphatase activity periodically (every 3–6 months) until it normalises, and then annually after stopping treatment. Relapse is common, often occurring many years after initial treatment, in which case further therapy should be offered. Relapse is usually apparent from an increase in alkaline phosphatase activity, but clinical relapse (recurrent pain) and radiological relapse (recurrence or progression of osteolytic lesions) can precede biochemical relapse. Bisphosphonate-induced osteonecrosis of the jaw is extremely rare when treating PDB, with an estimated risk of between one in 10 000 and one in 100 000.25
Nonpharmacological treatments should not be neglected. For example, patients with deformity resulting in leg shortening may benefit from shoe raises or orthotics to assist with pain relief and gait difficulties.
Summary
Paget disease is a common, treatable disorder affecting older Australians. Recent advances in therapy make long term remission a realistic goal in these patients.
Conflict of interest: John Walsh was a clinical investigator in two clinical trials of zoledronic acid for Paget disease.
References