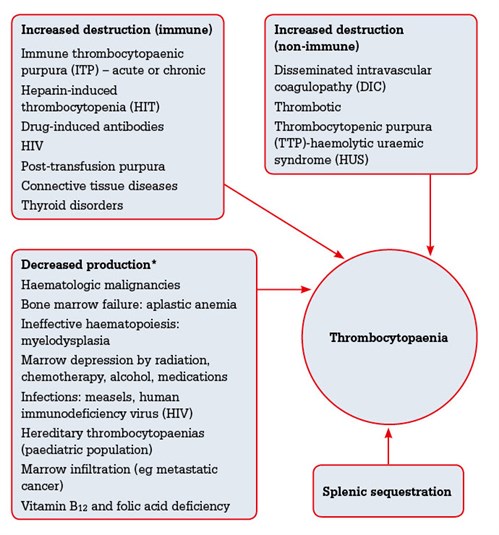
 |
Figure 1. Aetiology of thrombocytopaenia5
*Less likely to result in isolated thrombocytopenia |
Proposed approach to thrombocytopenia
Is it real?
Artefactual thrombocytopenia is found in about 1 in 1000 blood test results and can be a result of platelet aggregation, collection errors and/or platelet satellitism. An unexpected result, therefore, warrants a repeat test.3 In vitro clumping of platelets can occur when EDTA is used as an anticoagulant in the test tubes used for blood collection. This is thought to result from alteration of the platelet surface glycoproteins when incubated with a calcium chelator such as EDTA. As neither citrate nor heparins produce this phenomenon when used as anticoagulants, simultaneous collection of repeat blood in EDTA (used for full blood evaluation) as well as citrated test tubes (coagulation tubes) will confirm pseudo-thrombocytopenia.4 Alternatively, the platelet count can be determined by the laboratory haematologist using automated CD61 immunoplatelet analysis. A peripheral blood film is also vital in ruling out the possibility of clots for alternative reasons.
Is it new?
The previous blood results should be reassessed. A low platelet count may be normal for the patient and not represent illness. By definition, reference ranges would encompass only 95% of the population.5 In addition, a chronically mildly low platelet count (50–100 x 109/L) in a stable/asymptomatic patient with no other cytopaenias could be safely monitored. For example, slowly reducing platelet counts in a patient with chronic liver disease does not warrant an urgent referral.
Is it isolated?
Confirm that thrombocytopenia is isolated (ie. the full blood evaluation is otherwise normal and there are no red or white blood cell abnormalities on a peripheral blood film. An abnormal blood film with the presence of blast cells and/or any dysplastic changes warrants urgent referral (Table 1).
Table 1. Red flags
|
Red flags
|
Differential diagnoses
|
Clinical/laboratory findings
|
Management
|
|---|
|
Presence of bleeding
|
- Thrombocytopenia due to any cause
- Coagulation disorder
|
Mucosal or cutaneous bleeding
Easy bruising, profuse bleeding from superficial trauma, menorrhagia, metorrhagia, or melena
|
Urgent haematologist/emergency department referral
|
|
Presence of blasts on peripheral blood film
|
- Haematological malignancy
|
May be none
Lymphadenopathy and/or splenomegaly raise suspicion
|
Urgent haematologist referral
|
|
Evidence of haemolysis with the presence of neurological findings and renal dysfunction
|
|
The classic pentad:
- (MAHA)
- thrombocytopenia
- fever
- neurological findings*
- renal dysfunction**
(all five of these may not always be present).
Haemolysis screen includes:
- FBE and film
- reticulocyte count
- direct and indirect antibody test (Coombs)
- lactate dehydrogenase
- haptoglobin
- urinary haemosiderin
- haemoglobin11,34
HUS most commonly occurs after colitis resulting from Escherichia coli O157:H735 |
Early diagnosis and referral for plasmapheresis
|
|
Recent use of heparin or presence of skin necrosis on examination
|
|
HIT screen:
FBE
heparin-dependent platelet antibody, including assessing for cross-reactivity of the patients antibody with heparins and heparinoids11
Higher risk of HIT with previous exposure to unfractionated heparin, recent surgery and if the patient is female36
|
Cease heparin and avoid subsequent heparin therapy
|
|
Associated coagulation abnormalities
|
|
Platelets, fibrinogen and coagulation factors are being consumed. Hence these would be reduced
D-dimer, a marker for clot breakdown, would be elevated along with INR and PTT33
|
Urgent referral to the emergency department
|
|
Age >60 years and dysplastic features on peripheral film
|
|
More common for patients to present with anaemia and/or leukopaenia, but isolated thrombocytopenia may be the sole manifestation.
|
Referral of patients >60 years to a haematologist may be warranted
|
|
*Neurological signs: seizures, headaches, blurred vision, ataxia, change in mental status/fluctuating focal signs (ie motor deficits, diplopia, or aphasia)37
**Reduced urinary output or serum creatinine rise
DIC, disseminated intravascular coagulopathy; FBE, full blood evaluation; HIT, heparin-induced thrombocytopenia; HUS, haemolytic uraemic syndrome; INR, international normalised ratio; MAHA, microangiopathic haemolytic anaemia; MDS, myelodysplastic disorder; PTT, partical thromboplastin time; TTP, thrombotic thrombocytopenia
|
|---|
Is the patient pregnant?
Thrombocytopenia in pregnancy deserves special consideration because, rarely, it may have disastrous consequences for the fetus.6 Gestational thrombocytopenia is a common cause of mild thrombocytopenia (100–150 x 109/L). It is seen in up to 9% of pregnancies but must be differentiated from immune-mediated thrombocytopenia, which has a similar presentation (Table 2).6,7,9 At this stage, however, there is little information in the literature to guide the frequency of platelet counts in such patients. Non-urgent review by a haematologist is prudent.
Table 2. General guidelines for thrombocytopenia in pregnancy
|
Gestational thrombocytopenia
|
Immune thrombocytopenia
|
|---|
- No past history of thrombocytopenia
- Asymptomatic
- Mild (platelet count (100–150 x 109/L)
- Occurs late in the pregnancy or at term
- Not associated with significant bleeding risk
- No association with fetal thrombocytopenia
- Self-resolves following delivery
|
- Occurs early in the pregnancy
- May have a history of chronically low platelets
- Low platelet count (<50 x 109/L)
- Severe thrombocytopenia may develop in the infant in the first few days following delivery
|
It is important to keep in mind that serious obstetric emergencies such as the HELLP syndrome (haemolysis, elevated liver enzymes and low platelets) and disseminated intravascular coagulopathy (DIC), may be underlying, albeit rare, causes of thrombocytopenia (Table 1).8
Medication review
There is a wide range of medications associated with thrombocytopenia (Table 3). Although the most common presentation of drug-induced thrombocytopenia is severe symptomatic haemorrhage, an abnormal platelet count may be the first evidence of potential problems. Changes in medications over the past 10 days should be assessed. Normally it takes at least 5 days of drug exposure to develop thrombocytopenia, but as little as a few hours may be all that is required for fibrans and abciximab, or if the patient has been exposed to the drug previously.10
Table 3. Medications/mechanisms associated with thrombocytopenia10
- Immune-mediated mechanism (eg quinine, consumed through tonic water)
- Drug–hapten-induced haemolysis (eg penicillin, cephalosporins, tetracytcline, tolbutamide, semi-synthetic penicillins)
- Fiban-dependent antibodies (eg tirofiban)
- Monoclonal antibodies (eg abciximab)
- Autoantibody formation (eg gold)
- Immune complex formation (eg heparin)
|
|
The most commonly implicated drugs are quinine, quinidine, trimethoprim/sulfamethoxazole, vancomycin and chemotherapy drugs10
|
|---|
The gold standard for confirming this diagnosis is a drug re-challenge but this is rather impractical. A common pitfall is to refer to a haematologist without ceasing the suspected medications. As a general rule, cease the drug if suspicious. Investigations such as an indirect Coombs test, ELISA and flow cytometry may aid in the diagnosis and can be discussed with the laboratory haematologist.10,11
Presence of risk factors for chronic liver disease
Thrombocytopenia may be seen in up to 76% of patients with chronic liver disease (CLD).12 If there is clinical suspicion of high alcohol use, hepatitis, intrinsic liver disease or obstruction, clinical assessment for stigmata of CLD followed by formal liver function tests are warranted. Further assessment of cirrhosis by ultrasonography or other modality may also be indicated, but an urgent referral to the haematologist may not be required.
Distributional thrombocytopenia
The spleen normally sequesters 30–35% of the body’s platelets, but this can rise to 80–90% when it is enlarged, causing thrombocytopenia.13 Routine assessment of splenomegaly on clinical examination is important.
Concurrent B12 and folic acid deficiency
About 20% of patients with megaloblastic anaemia due to vitamin B12 and folic acid deficiency also have thrombocytopenia. These deficiencies should be suspected in patients with excess alcohol use or malnourishment, and the elderly with poor oral intake.14,15
Risk factors for HIV, hepatitis C and/or recent infections or live vaccinations
Infections or live vaccinations may cause thrombocytopenia by direct bone marrow suppression or increased peripheral consumption. An isolated low platelet count may be the initial manifestation of disease in as many as 10% of patients with HIV.16,17 Some of the other commonly associated viruses include Epstein-Barr virus, cytomegalovirus, rubella virus, parvovirus B19 and mumps.18
Suspected systemic autoimmune disorder
There is an association between systemic autoimmune disorders such as systemic lupus erythematosus (SLE) and the antiphospholipid syndrome and thrombocytopenia.19 One study found that 29 out of 50 patients (58%) diagnosed with SLE had thrombocytopenia at the time of diagnosis and this was associated with higher morbidity, affecting overall prognosis.20 Thus, investigations for antibodies may be warranted in the presence of suggestive symptoms; however, routine testing does not need to be performed as there is no gold standard diagnostic test. Additionally, false positives as well as weak true positive results are not uncommon.21
Consider congenital thrombocytopenia
Congenital conditions that cause thrombocytopenia are quite heterogenous and uncommon. They usually present in the paediatric age group with bleeding diathesis. Mild cases, however, may remain undiagnosed until older age. This group may remain completely asymptomatic or may have only sporadic thrombocytopenia during times of haemostatic stress (eg. surgery, trauma, etc); careful history taking is key.
Previous history of easy bruising, prolonged bleeding or petechiae and a family history raise suspicion. The diagnosis may be narrowed further if ‘giant platelets’ or microthrombytes are found in a peripheral blood smear.22 If congenital aetiology is highly suspected, a haematologist referral is indicated.
Immune thrombocytopenic purpura (ITP)
ITP is an immune disorder characterised by isolated thrombocytopenia and is strictly a diagnosis of exclusion. The incidence is estimated at approximately 1.6–3.9 per 100,000 person years.23 It is caused by increased platelet clearance and decreased production, but the details of how these develop are complex and for the most part remain undetermined.
The primary treatment goal is to prevent severe bleeding rather than achieve normal platelet counts. In general, almost half of the new diagnoses of primary ITP do not require treatment and there is also no gold standard investigation.25 Response to ITP-specific therapy is supportive of the diagnosis.26 It is important to mention that the prevalence of thyroid disorders is greater in people with ITP than the general population. Testing thyroid function in patients with ITP may be beneficial.
Red flags – when is immediate referral required?
Presence of blasts on a blood film
Presence of blast cells and/or any dysplastic changes is likely to be the first presentation of a haematological malignancy.27
Evidence of haemolysis with or without neurological findings and renal dysfunction
Thrombotic thrombocytopenic purpura (TTP) and haemolytic uraemic syndrome (HUS) are multi-system disorders characterised by thrombocytopenia and microangiopathic haemolytic anaemia (MAHA) (Table 1). TTP and HUS are life-threatening illnesses; TTP has a mortality rate of >90% without appropriate treatment.28
Recent use of heparin or presence of skin necrosis on examination
Heparin-induced thrombocytopenia (HIT) is a hypersensitivity reaction to unfractionated heparin and low molecular weight heparin mediated by an IgG antibody.29 One study found that the risk of HIT with low molecular weight heparin was lower (0.2%), compared with unfractionated heparin (2.6%).30 There are two forms of HIT, type I and type II. Type I HIT occurs more frequently and does not require cessation of therapy. Type II is immune-mediated and can cause venous or arterial thrombosis, which may be fatal or require limb amputation. In general, about half of the patients with HIT also have thrombosis.31 In a critical situation, there is no test that can be performed with sufficient speed, sensitivity and specificity; therefore, the diagnosis is primarily clinical.31 This is a medical emergency. The in-patient mortality for HIT is up to 20%.32
Associated coagulation abnormalities
Acute or chronic disseminated intravascular coagulopathy is a consumptive coagulopathy associated with end-organ damage. It may present with a background of many conditions including pregnancy, malignancies and systemic infections.33 Essentially clotting and clot breakdown are occurring simultaneously. There is no single laboratory test for diagnosis (Table 4).
Table 4. Investigations for thrombocytopenia in an asymptomatic patient
|
First-line investigations
|
Not routine/ordered by specialist
|
|---|
|
FBE and peripheral blood film
Platelet indices (size and volume): limited value
Fibrinogen, D-dimer, clotting factors
Haemolysis screen (see Table 3)
Vitamin B12 and folic acid levels
Liver function tests
HIV and hepatitis serology1
Stool for occult blood
Renal function (deranged in TTP/HUS)
|
Anti-platelet antibody test39
Tests for SLE, anti-phospholipid syndrome
Quantitative immunoglobulin level
Bone marrow biopsy
Helicobacter pylori (ITP refractory to treatment)
Thyroid function tests
|
|
FBE, full blood evaluation; HUS, haemolytic uraemic syndrome; ITP, immune thrombocytopenic purpura; SLE, systemic lupus erythematosus; TTP, thrombotic thrombocytopenic purpura
|
|---|
Older age (>60 years) and dysplastic features on peripheral film
Myelodysplasia describes the abnormal development of one or more of the three major haematopoietic lineages (ie. red blood cells, while blood cells, platelets). These are pre-leukaemic disorders, more common in people older than 60 years and warrant particular attention as they may manifest solely as isolated thrombocytopenia with subtle changes on peripheral film. They should be considered in all patients >60 years of age with progressive or significant thrombocytopenia (<50 x 109/L). As a guide, bone marrow tests are appropriate people aged over 60 years who have thrombocytopenia but are otherwise asymptomatic.1
Suggested management in the absence of red flags
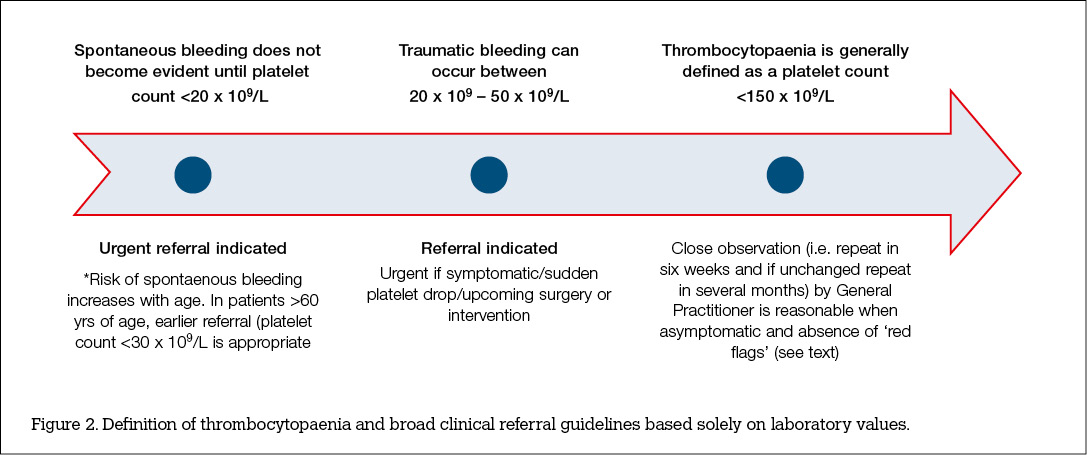
After excluding red flags and other common causes listed above, patients with modest decreases (50–100 x 109/L) but are asymptomatic may have a repeat test in 1–2 weeks. Broad clinical referral guidelines based on the ranges of laboratory values can be useful (Figure 2, available online only). With minimum degrees of asymptomatic thrombocytopenia (100–150 x 109/L), the tests may be repeated in 1 or more months.17 Patients with symptomatic thrombocytopenia but otherwise healthy may require no activity restriction, but all patients need to be advised regarding symptoms and signs suggestive of severe thrombocytopenia, such as mucosal bleeding, petechiae and easy bruising.
Key points
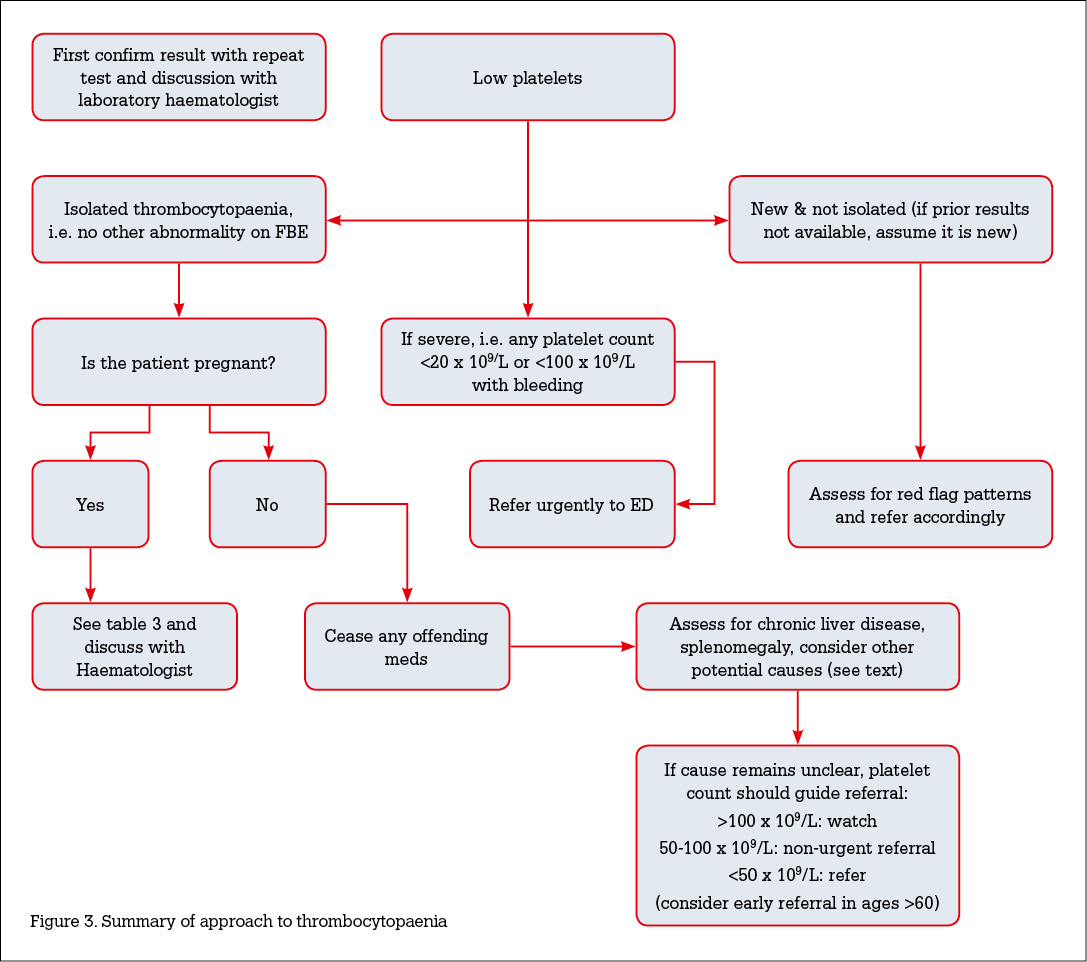
- Thrombocytopenia is a common finding and has a wide range of aetiologies. A systematic approach to identifying the underlying causes is essential (Figure 3, available online only).
- Due to lack of specific tests, careful history, examination and blood evaluation remain the initial means of diagnosis.
- A list of common benign causes of isolated thrombocytopenia and red flags can help the general practitioner triage those at high risk.
Competing Interests: Hannah Rose received an honorarium from Amgen for the development and presentation of lectures to nurses in May 2014.
She has also been provided with accommodation by Novartis and Roche for attendance at educational meetings in Melbourne over the past 12 months, although no money was received directly in relation to this.
Provenance and peer review: Not commissioned; externally peer reviewed.