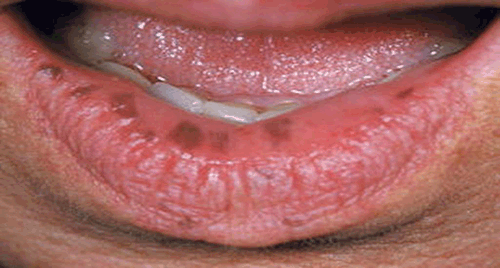
Figure 1. The patient’s lower lip
Question 1
What is the most likely diagnosis?
Question 2
What other differential diagnoses should you consider?
Question 3
What is the significance of these lesions?
Question 4
What laboratory investigations should be ordered?
Question 5
What further management is advised?
Answer 1
The most likely diagnosis is Peutz-Jeghers syndrome (PJS).
Answer 2
Differential diagnoses for oral hyperpigmented lesions should include:
- oral naevi and melanotic (melanocytic) macule: usually solitary and remain unchanged in size and colour
- oral melanoacanthoma: precipitated by a traumatic event and associated with inflammation and spontaneous resolution
- smoker’s melanosis: very similar appearance (plus history of smoking)
- Addison disease: similar oral lesions (plus generalised hyperpigmentation, weight loss and hypotension).
Answer 3
The presence of hyperpigmented oral macules, and the strong family history of gastrointestinal malignancies in both her mother and brother, is suggestive of Peutz-Jeghers syndrome.1 Peutz-Jeghers syndrome is an inherited condition, whose key features are mucocutaneous hyperpigmented macules in association with hamartomatous polyps in the gastrointestinal tract. The cause is a specific germline alteration (usually deletion) at the serine threonine kinase 11 (STK11) gene on chromosome 19. This can arise de novo or through autosomal dominant inheritance.2 The condition is very rare, occurring in 1 in 25 000 to 300 000 births.
The STK11 is thought to be a tumour suppressor gene. Hence, patients with PJS are significantly more likely to develop cancers, especially in the gastrointestinal tract, breasts and pancreas.3,4
Answer 4
Laboratory investigations include an annual full blood examination (FBE), as the hamartomatous polyps can ulcerate and bleed leading to iron deficiency anaemia. If there is microcytic, hypochromic anaemia, iron studies should be carried out. Faecal occult blood can also be performed to check for blood in the stool.
Genetic testing can be offered to the patient. The latest genetic test is the multiplex ligation probe amplification (MLPA) assay.
Answer 5
The patient should be counselled and educated regarding the symptoms and potential complications of gastrointestinal bleeding, intestinal obstruction and increased risk of malignancy. The importance of genetic screening, disease monitoring and cancer surveillance should also be emphasised for both the patient and her immediate family.
Cancer surveillance
Patients with PJS have up to a 93% lifetime risk of developing cancer3,4 in the liver, lungs, breasts, ovaries, uterus, and testicles. For patients with PJS, the relative risk for all cancers was 15.2 compared to the general population, between the ages of 15 and 64 years.3
Recommendations for review and cancer screening for people with PJS have included:
- annual for all: history, physical examination, FBE5
- annual depending on age: ultrasonography of the pancreas and pelvis, mammogram (or magnetic resonance imaging [MRI] breast) and Pap test5
- biennial:1,6 endoscopic surveillance to remove polyps and reduce the risk of obstruction, bleeding and cancer. For confirmed PJS patients, upper gastrointestinal tract endoscopy and capsule endoscopy7 for fully evaluating the small bowel should start from 10 years of age, or younger if symptomatic. Magnetic resonance imaging enterography may prove to be an appropriate alternative.8 Colonoscopy should be performed from the age of 25 years.
Chemoprevention
The COX-2 enzyme is over-expressed in patients with PJS. Limited evidence shows that COX-2 inhibition with celecoxib can suppress polyp growth,9 but there is an increased risk of myocardial infarct and stroke. Hence, there is no recommendation as yet for its use in this condition.
Genetic counselling and screening
As this condition is autosomal dominant, patients and their families should be referred to specialist family cancer clinics for genetic counselling, screening and coordination of management.10