This article is the first in a series on pathology testing. Articles in this series aim to provide information about emerging laboratory tests that general practitioners may encounter.
Prenatal screening for fetal chromosomal abnormalities is carried out to identify women who are at higher risk of having an affected fetus. It also enables them to make informed decisions about whether to proceed to diagnostic testing.1 Clinically significant fetal chromosomal abnormalities generally involve gains or losses of genetic material. These can range in size from small segments of chromosomes (termed ‘microduplications’ or ‘microdeletions’) to entire chromosomes (ie aneuploidy).2
The most common chromosomal abnormality is trisomy 21 (ie presence of an additional copy of chromosome 21), which causes Down syndrome. Other fetal aneuploidies are generally associated with spontaneous pregnancy loss, but some, particularly trisomy 18 and 13, can result in live births. Most cases of trisomy 21, 18 and 13 arise de novo (as a spontaneous event), although in rare cases there may be a predisposing parental chromosomal rearrangement, such as a translocation. The likelihood of each of these three aneuploidies increases with maternal age.3
In Australia, the most common screening modality for fetal chromosomal abnormalities is the combined first-trimester screen (cFTS).1 This is carried out between 11+0 and 13+6 weeks of gestation, and combines ultrasound measurements, including nuchal translucency, maternal serum analytes (human chorionic gonadotropin [hCG], oestradiol, pregnancy-associated plasma protein A [PAPP-A]) and maternal age to produce a risk score.1,4 If the risk score is higher than a given cut-off value, it is considered a ‘screen-positive’ or ‘high-risk’ result, indicating that diagnostic testing should be considered.
Diagnostic testing requires an invasive procedure. This can be carried out between 11 and 14 weeks of gestation by chorionic villous sampling (CVS) of placental tissue. Alternatively, after 15 weeks of gestation, fetal amniocytes can be sampled by amniocentesis.5 Both procedures carry a small risk of provoking spontaneous miscarriage. The degree of risk is commonly quoted as 0.5–1%, although recent meta-analyses suggest that the true procedure-related risk may be much lower.6 Chromosomal abnormalities can be diagnosed in cells derived from the invasive procedure by karyotyping or at higher resolution by microarray analysis.2
Non-invasive prenatal testing – Cell-free DNA
‘Cell-free’ DNA (cfDNA) consists of short DNA fragments, which are released into plasma from normal cellular turnover and are rapidly cleared from circulation. In a woman who is pregnant, most of the cfDNA is derived from turnover of maternal cells. However, a proportion is derived from the outer trophoblast cell layer of the placenta, which typically reflects the fetal genotype.7 The percentage of cfDNA derived from the trophoblast is termed the ‘fetal fraction’. There is a wide normal range of fetal fraction. The median value at 10 weeks of gestation is approximately 10%.8
Non-invasive prenatal testing (NIPT) tests differ in their exact methodology and there are several different assays available in Australia; a detailed comparison is beyond the scope of this review. In general, NIPT assays examine the proportion of cfDNA derived from specific chromosomes. Fetal aneuploidy can cause these proportions to deviate from expected values, and statistical tests are applied to determine whether such deviations are significant.9,10 As the majority of cfDNA is maternal, the ability to detect an abnormality of a given fetal chromosome requires sufficient fetal fraction. Many NIPT assays therefore have a fetal fraction cut-off level, and samples with fetal fraction below the defined cut-off do not produce a result.8,11,12
NIPT can be carried out at any point in the pregnancy from 10 weeks of gestation onwards to increase the likelihood of sufficient fetal fraction. NIPT typically requires a specific request form, and can be requested by a medical practitioner (general practitioner or obstetrician) who is involved in the patient’s antenatal care.
NIPT compared with cFTS
Using the cFTS, a detection rate of approximately 85–90% can be achieved for trisomy 21, 18 and 13, at a false positive rate of 4–5%.4,13 In a pooled meta-analysis, the detection rate across different NIPT methods was just over 99% for trisomy 21, 96% for trisomy 18 and 91% for trisomy 13. The cumulative false positive rate was less than 0.4%.14
In addition to detection and false positive rates, the positive and negative predictive values (PPV and NPV) of a screening test are important clinical parameters. These values depend partly on the performance characteristics of the test, but also vary with the prevalence of the tested condition in the population.15 Low prevalence of a condition will decrease the PPV and increase the NPV of a screening test, whereas high prevalence will have the opposite effect.
Predicted PPV and NPV can be modelled for trisomy 21, assuming detection rates of 99% and 90% and false positive rates of 0.1% and 4% for NIPT and cFTS respectively. Table 1 shows the PPV of a high-risk screening result and NPV of a low-risk screening result for three groups with different prior risk levels.
Table 1. The PPV and NPV of cFTS and NIPT depend on prior risk
|
Prior risk
|
PPV of cFTS
|
PPV of NIPT
|
NPV of cFTS
|
NPV of NIPT
|
|---|
|
1 in 4 (very high risk)
|
88.2%
|
99.7%
|
96.6%
|
99.7%
|
|
1 in 300 (common cFTS cut-off for invasive testing)
|
7.0%
|
76.8%
|
>99.9%
|
>99.9%
|
|
1 in 950 (risk for a 25-year-old at 12-week gestation)
|
2.3%
|
51.1%
|
>99.9%
|
>99.9%
|
|
cFTS, combined first trimester screen; NIPT, non-invasive prenatal testing; NPV, negative predictive value; PPV, positive predictive value
|
As Table 1 shows, the PPV of NIPT is never 100%9,10 and NIPT is therefore a screening test. Following a high-risk result, invasive diagnostic testing is required to provide certainty regarding fetal genotype and is strongly recommended if a patient is considering termination of pregnancy.1,16–18 Similarly, a low-risk NIPT result does not guarantee absence of the screened abnormalities, particularly if the patient’s prior likelihood is very high.
Differences between the NIPT result and fetal genotype may arise for technical reasons; for example, because NIPT is based on counting statistics, there will be a small number of statistical outliers. There are also several potential biological reasons for either false negative or false positive NIPT results,9,10 including the following:
- A low fetal fraction can potentially lead to a false negative result. This is more common in patients with a high body mass index.8,11,12
- In the case of twins, surviving placenta from a demised twin can release cfDNA, leading to a false positive result (or in theory, a false negative result). Note that most NIPT assays are validated for twin pregnancies, although the test failure rate is higher in twins and the detection rate may be lower.14 Higher order multiples are not generally tested by NIPT.
- Maternal chromosomal abnormalities can lead to a false positive result. Examples include mosaic constitutional chromosomal abnormalities or copy-number variants, presence of a bone marrow or tissue transplant and, in rare cases, maternal malignancy.
- Different genotype of the fetus and the placental trophoblast, either ‘confined placental mosaicism’ or ‘true-fetal mosaicism’ with feto-placental discordance, can lead to a false positive or false negative result respectively.19
- NIPT may not detect rare mosaic or partial trisomies of the targeted chromosomes.9,14,16–18
Complexities of NIPT
NIPT is an effective screening modality for targeted chromosomal abnormalities, but it is important to be aware of associated complexities, which should be considered in pre-test counselling.
Low fetal fraction and assay failures
A proportion of NIPT samples fail to produce an interpretable result. This can be for a variety of reasons, including low fetal fraction, specimen-related issues or suboptimal data quality.10,12,14 The reported failure rate varies among NIPT assays, ranging from 1.6% to 6.4%.17,20
Fetal fraction increases with gestational age and correlates inversely with maternal weight.8,11,20 There is emerging evidence suggesting that fetal fraction can be altered by other maternal or placental factors.21 Fetal fraction appears to be lower in the presence of certain fetal chromosomal abnormalities, particularly trisomy 13 and 18, monosomy X, and triploidy. There may therefore be an increased risk of these abnormalities in samples that fail NIPT because of low fetal fraction, although the degree of this risk has not been fully established.12,17,20
The American College of Medical Genetics and Genomics therefore recommends that invasive testing should be offered following failed NIPT.18 An alternative approach might be to interpret failed NIPT in the context of other risk indicators, such as cFTS risk score and detailed ultrasonographic findings.12 If other factors indicate high risk of a chromosomal abnormality, invasive testing may be the best option. For low-risk patients, particularly if they are early in gestation, repeating NIPT may be considered. Repeat testing produces a result in approximately two-thirds of patients.12,20
Which chromosomal abnormalities should be screened for?
In addition to trisomy 21, 18 and 13, many NIPT providers offer additional screening for sex chromosome aneuploidies. In general, the clinical presentation of sex chromosome aneuploidies is less severe and more variable than autosomal aneuploidies. The NIPT pooled detection rate is approximately 90% and the false positive rate is approximately 0.4%.14 PPV is typically lower than for autosomal aneuploidy, mainly because of confined placental mosaicism or maternal sex chromosome abnormalities. Considering these factors, most guidelines advise careful pre-test counselling for patients contemplating screening for sex chromosome aneuploidy.1,16–18
Some NIPT providers also offer screening for a panel of selected microdeletions. Individual microdeletions can present with variable clinical phenotypes and are much rarer than autosomal aneuploidies. There is, therefore, less analytical validation and clinical trial data regarding NIPT performance for microdeletions. In addition, the rarity of these abnormalities means that the PPV will be lower than for autosomal or sex chromosome aneuploidies, potentially leading to an increased rate of invasive testing and eroding a key benefit of NIPT.
Current guidelines are divided regarding microdeletions. Some state that screening should not be offered routinely,1,16 whereas others suggest screening can be offered after careful pre‑test counselling, with invasive testing and microarray a consideration for patients who are particularly concerned about these abnormalities.17,18
Which chromosomal abnormalities are not addressed by NIPT?
Trisomy 21, 18, 13 and sex-chromosome aneuploidies make up the majority of chromosomal abnormalities detectable by karyotype after invasive testing. However, there are a number of other chromosomal abnormalities that are individually rare, but collectively are relatively common. These ‘atypical’ abnormalities may cause fetal structural abnormalities or alter cFTS parameters such as nuchal translucency or maternal serum markers.5
Atypical abnormalities appear to be enriched in patients with high-risk scores on the cFTS. In patients with high-risk scores on conventional screening tests, 20–30% of potentially significant chromosomal abnormalities may be undetectable by NIPT. The residual risk of atypical abnormalities after a low-risk NIPT result in this group may be 1–2%.22,23 If microdeletions and microduplications detectable by prenatal microarray are also considered, the proportion of potentially significant abnormalities detectable by NIPT is further reduced.24
Finally, NIPT cannot currently detect single-gene disorders such as Fragile X syndrome or cystic fibrosis, or non‑genetic abnormalities such as neural tube defects or congenital cardiac anomalies.9,16–18
Clinical application of NIPT
The major clinical benefit of NIPT is to increase the detection rate for the targeted abnormalities, while simultaneously reducing the number of false positive results and invasive tests. However, given its complexities and costs, the optimal use of NIPT remains a subject of debate. In the Australian context, there is currently no Medicare Benefits Schedule (MBS) rebate for NIPT and test costs are in the range of $400–$500 dollars.
The Royal Australian and New Zealand College of Obstetricians and Gynaecologists (RANZCOG) guidelines acknowledge NIPT as an option for pregnant women, but do not offer prescriptive algorithms for its integration into prenatal care.1 On the basis of these and other guidelines, two possible pathways for clinical use of NIPT are presented for women who choose to undergo antenatal screening for fetal chromosomal abnormalities (Figure 1). The first is the ‘contingent model’, where use of NIPT is triaged after an initial cFTS result; the second is the ‘NIPT first’ model.1,16–18,25
In both models, if NIPT fails, the appropriate action should be considered in the light of the patient’s wishes, the gestation of the pregnancy and other risk factors for fetal chromosomal abnormality. Detailed ultrasonography and cFTS or second-trimester screening may prove useful to clarify risk. For some patients, it may be appropriate to offer invasive testing after NIPT failure. For others, repeating NIPT may be appropriate – for example, if they are at low risk on the basis of other parameters and/or if they are early in gestation.12,18,20
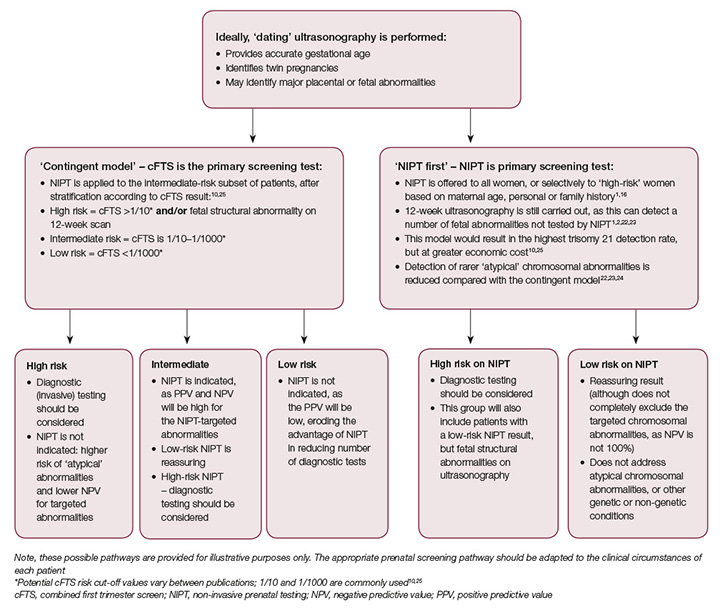
Figure 1. Possible pathways for use of NIPT in clinical practice
Summary
NIPT is a screening test carried out on circulating trophoblast-derived cfDNA after 10 weeks of gestation. Its primary utility lies in increasing the detection rate for targeted aneuploidies, while reducing the rate of invasive testing.
As NIPT is a screening test, the possibility of false positive or false negative results for the targeted abnormalities should always be considered. NIPT results should be interpreted in the light of all available information about the pregnancy. Confirmation of high-risk results by invasive testing should be carried out before making irreversible decisions about the pregnancy.
NIPT has complexities around appropriate targeted abnormalities, the residual risk of atypical chromosomal abnormalities and test failures. In addition, there is no single optimal protocol for clinical use. Despite this, it offers clear benefits in terms of PPV and NPV for the targeted chromosomes, and should be considered as an option for pregnant patients.1
Author
James Harraway MBChB, FRCPA, DPhil, Genetic Pathologist, Sullivan Nicolaides Pathology and Mater Pathology, Qld. james_harraway@snp.com.au
Competing interests: Dr Harraway supervises NIPT using the Harmony Prenatal Test (Roche Diagnostics) at Sullivan Nicolaides Pathology.
Provenance and peer review: Commissioned, externally peer reviewed.
Acknowledgments
I would like to acknowledge the contribution of Professor Graeme Suthers and Dr Victoria Harraway to the drafting and review of this article.