Growth is a fundamental process of adolescence, and growth issues often have profound effects on the young person. This article will provide a guide to assessing growth issues in this age group, and a framework for investigations and when to consider referral.
Normal growth
There are three phases of growth:
- infancy, which is nutritionally driven and lasts until approximately 24 months of age
- childhood, when the growth hormone (GH)–insulin-like growth factor 1 (IGF-1) axis takes over as the major regulator of linear growth
- adolescence, when sex steroids and puberty drive growth and bone mass accrual between 10 and 19 years of age.1
This article will focus on growth disorders in adolescence.
Measuring height of an adolescent
Height should be measured with a wall-mounted stadiometer when the patient is not wearing shoes, has their ankles together and gaze looking forward. A gentle upward pressure on the mastoid processes helps to achieve a ‘true height’.2 A child’s height must be interpreted within the context of their mid-parental height, which should be charted on the right-hand side of a child’s gender-specific growth chart. It is commonly held that a person will reach within 10 cm of the mid-parental height expectation.3 There are several ways to calculate mid-parental height, but two easy formulas to remember are:
- Tanner method:
- Girls: [(father’s height in cm + mother’s height in cm)/2] – 6.5
- Boys: [(father’s height in cm + mother’s height in cm)/2] + 6.5
- Alternatively:
- Girls: father’s height in cm – 13
- Boys: mother’s height in cm + 13.
Predicting the final adult height becomes less accurate when parents are at opposite extremes of height.3
Effect of normal puberty on growth
Puberty is a complex and intricate cascade of events, requiring gonadal and adrenal awakening. Tanner staging is widely used to describe pubertal stages, with correlating pictorial descriptions of all five stages (Figure 1). Thelarche (palpable subareolar breast buds), under the influence of oestrogen, is usually the first marker of puberty in girls. This generally occurs around 10 years of age, with a normal age range of 8–13 years, and is followed by menarche, generally 2–2.5 years later.1,4–7 For boys, an increase in testicular volume of 4 mL or greater is generally the first sign of pubertal development. This increase in volume is due to growth of seminiferous tubules under the influence of follicle-stimulating hormone (FSH), and generally occurs around 11 years of age, with a normal age range of 9–14 years.1,4–7 Testicular volume is most commonly measured using a Prader orchidometer.
From the age of four years and throughout childhood, the average growth velocity is 5–7 cm per year. During puberty, axial skeletal growth accelerates and a greater proportion of trunk length is gained.8 Growth velocity in puberty increases to approximately 6–10 cm per year for girls and 7–12 cm per year for boys.1,9,10 Peak growth velocity occurs just after thelarche (around Tanner breast stage 3), but prior to the onset of menses for girls (around 11.5−12 years of age); and around 13–14 years of age for boys. Given that puberty – and hence peak growth – occurs later in boys, coupled with their higher peak growth velocity, the difference between final height is approximately 12.5 cm between the sexes.
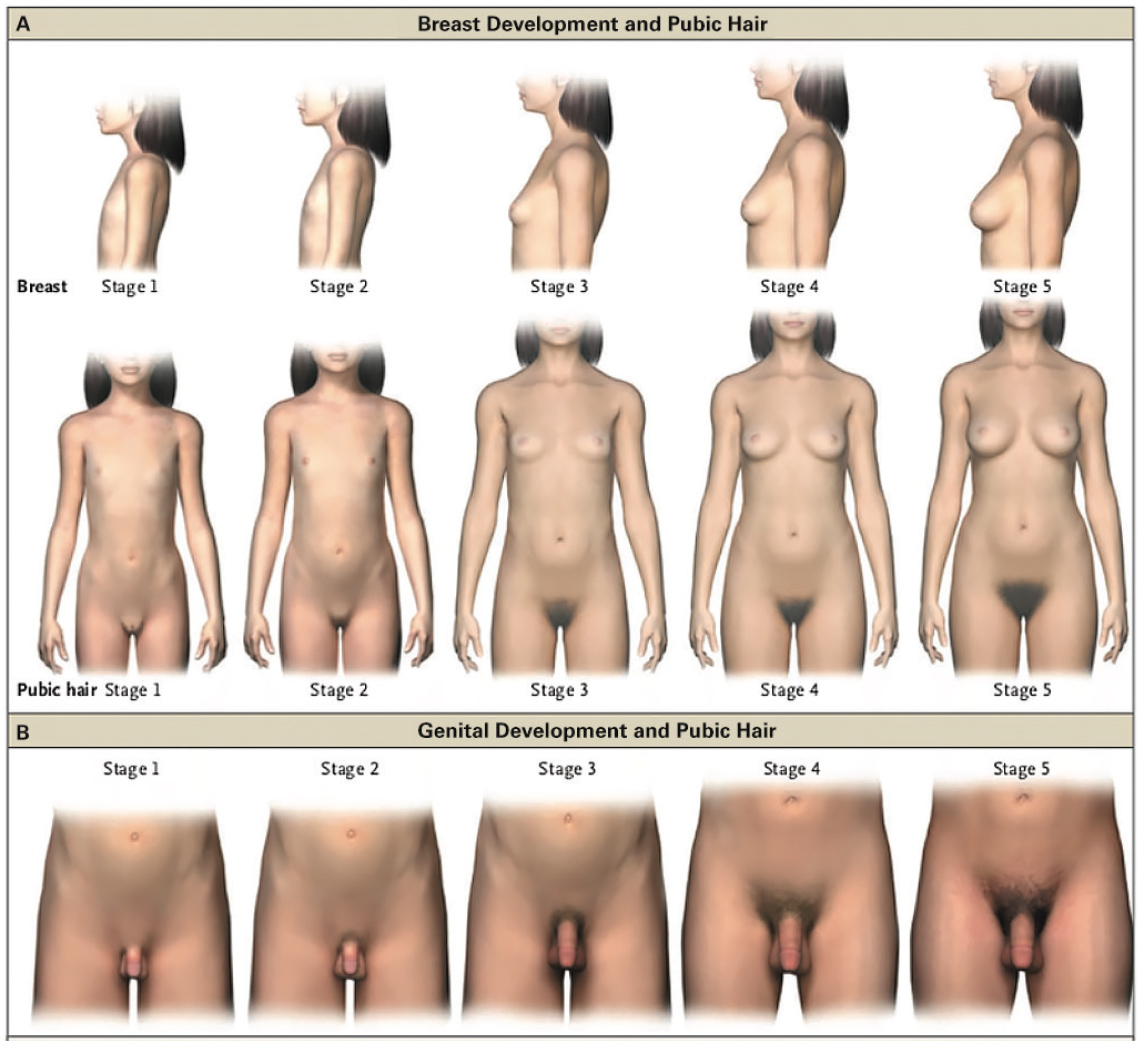
Figure 1. Pubertal Rating according to Tanner stages
Reproduced with permission from NEJM Group from Carel JC, Léger J. Clinical practice. Precocious Puberty. N Engl J Med 2008;358(22):2366–77.
When to suspect a growth disorder in an adolescent
Once mid-parental height has been plotted, growth velocity must also be assessed. Multiple anthropomorphic measurements will give a more accurate indication of growth velocity.2 Following this, a detailed history and examination are required (Box 1).
Box 1. History and examination
|
Features on history
- Birth – growth in utero, birth weight, gestation, maternal factors (gestational diabetes, smoking, alcohol intake)
- Neonatal period – floppiness, hypoglycaemia, feeding issues
- Detailed past medical history – prolonged illness or hospitalisation, chronic or systemic illnesses, medications, developmental progress
- Full systems review to exclude systemic disease
- Dietary history – malnutrition, anorexia nervosa
- Family history – consanguineous parents, parental and sibling heights, pubertal timing in relatives
Examination findings
- General examination – include specific systems examinations for conditions elicited on history, dysmporphism
- Body proportions – arm span, upper limb and lower limb segments
- Height
- Weight on calibrated scales
- Pubertal assessment as above
- Visual field examination, assessment of smell – simple bedside tests that can alert to possible intracranial pathology
|
Short stature presenting in adolescents
Short stature is defined as height for age that is less than two standard deviations below the 50th centile for gender (ie height less than the third centile on standard growth charts).11 The most common causes of short stature are constitutional delay of growth and puberty (CDGP) and familial short stature (FSS; Table 1).11,12 In FSS, the child is short when compared with their peers, but they are an appropriate height for their family, as evidenced by their mid-parental height. Monitoring growth is generally all that is required.
The diagnosis of CDGP is based on a lack of breast development (Tanner stage 2) in girls aged >13 years, and testicular volume <4.0 mL by the age of 14 years in boys. The adolescent, male or female, may find the discrepancy in their height and lack of secondary sexual characteristics, compared with peers, distressing, which often prompts referral to the endocrinologist. An accurate family history may assist in making the diagnosis of CDGP, and will often elicit late onset of age of puberty in parents or older siblings. Inheritance of CDGP is thought to be in a dominant mode, with prevalence being significantly higher in boys, compared with girls (ratio of 3:1).13 However, the almost equal proportions of first-degree relatives with CDGP suggest that the higher number of boys may be the consequence of referral bias.13 This is likely to be multifactorial, possibly due to societal notions of height and more obvious initial signs of female puberty. Without intervention, most adolescents with CDGP will undergo spontaneous puberty. If the adolescent is experiencing emotional distress, then low-dose testosterone (intramuscular, oral, topical) has been used judiciously to induce puberty in males aged 14 years or older.14,15 There are no clear guidelines on when to use oestrogen in females with CDGP. It is important to highlight that in males over the age of 12 years and females over the age of 10 years, CDGP precludes the child from receiving GH therapy if their final height prediction, once corrected for bone age, is above the first centile for gender-specific growth charts.16
Table 1. Common forms of short and tall stature
|
|
Chronic disease
|
Constitutional delay of growth and puberty
|
Familial short stature
|
Familial tall stature
|
|---|
|
Height
|
Short
|
Short
|
Short
|
Tall
|
|
Bone age
|
Possibly delayed
|
Delayed
|
Not delayed
|
Not advanced
|
|
Growth rate
|
Slow
|
Slow
|
Normal
|
Normal
|
|
Height prognosis
|
Depends on control of underlying condition
|
Generally good
|
In keeping with genetic potential
|
Good
|
Turner syndrome
Turner syndrome (45 XO karyotype or mosaic variants) is a common syndromic cause of short stature, with an incidence of one in 2000. Short stature in Turner syndrome is thought to be due in part to haploinsufficiency of the short stature homeobox (SHOX) gene, which is located on chromosome X. Even when ‘classical features’ are lacking, Turner syndrome should be considered in any girl with short stature, stature shorter than her mid-parental height expectation, or delayed or arrested puberty. Without treatment with GH, the average height for a woman with Turner syndrome is 136–147 cm. Even with GH therapy, outcomes vary because of the many different GH treatment regimens, the age of treatment commencement, and the highly heterogeneous nature of Turner syndrome.1,17,18
Acquired growth hormone deficiency
Primary endocrine causes for short stature presenting in adolescence are much less common and often have increased weight for height. These causes include acquired hypothyroidism, glucocorticoid excess (Cushing’s syndrome) and acquired GH deficiency (GHD). Acquired hypothyroidism can present with declining growth velocity and is often due to autoimmune thyroiditis in adolescence. Symptoms can be insidious and include lethargy, cold intolerance, constipation and poor school attention. Endogenous Cushing’s syndrome (central, peripheral) is rare in children; however, exogenous Cushing’s syndrome due to glucocorticoid therapy (ie high-dose inhaled or systemic) is much more common.19
Acquired GHD should be considered in any child who has had an intracranial tumour, cranial radiation or brain trauma. Signs that suggest acquired GHD may include proportional short stature, decline in growth velocity and symptoms secondary to the underlying cause, such as raised intracranial pressure and head injury. A serum IGF-1 level is used to screen for GHD; however, a single low IGF-1 level, even in the absence of poor nutrition, does not always represent GHD.1,20 Given the pulsatile secretion of GH, the diagnosis of GHD is made after provocation testing (eg with glucagon, arginine, clonidine). Children with true GHD have striking catch-up growth on recombinant human GH therapy, particularly in the first year of treatment.1,21 In cases of short stature without biochemical GHD (ie idiopathic short stature), the use of GH has produced a 3.5–7.5 cm increase in final adult height,22,23 although there is some evidence that commencement closer to the end of growth limits the expected response.24
Non-endocrine causes
There are many other non-endocrine causes of short stature. Importantly, antenatal conditions, such as extreme pre-term gestation and intrauterine growth restriction or low birth weight, have long-lasting effects on growth and final height.25 A detailed history may also reveal whether poor nutrition (eg malnutrition, anorexia nervosa) or extreme physical activity (eg gymnastics, ballet) are leading to maturational delay and possible growth slowing. Beyond this, chronic disease and/or inflammatory conditions (eg inflammatory bowel disease, chronic kidney disease, coeliac disease) can present with attenuated growth during childhood, often with a delayed bone age.12 Treatment side effects, particularly related to oncology sequelae (spinal radiation), exogenous glucocorticoids (inhaled and systemic), recurrent blood-product transfusions, or stimulant medications for behavioural disorders (eg attention deficit hyperactivity disorder) also need to be considered as possible causes of short stature. Refer to references 12 and 26 for further reading on short stature.
Growth hormone
In Australia, the use of Pharmaceutical Benefits Scheme (PBS)-subsidised GH is tightly regulated. The most common indication for GH use is ‘short-slow growing’, which is defined as height less than the first centile and growth velocity greater than the 25th centile for bone age. However, if the young person has significant maturational delay, it may be more appropriate to induce puberty rather than treat with GH. Box 2 lists the conditions for which PBS-funded GH is available in Australia.
Box 2. Conditions where PBS-subsidised GH may be available in Australia16
- Short stature and slow growth
- Biochemical growth hormone deficiency
- Turner syndrome or SHOX disorders
- Prader–Willi syndrome
- Growth retardation secondary to an intracranial lesion, or cranial irradiation (lesion needs to be stable for 12 months prior to application)
- Chronic renal insufficiency
|
|
GH, growth hormone; PBS, Pharmaceutical Benefits Scheme; SHOX, short stature homeobox
|
Tall stature presenting in adolescents
Tall stature is defined as height greater than the 97th centile on standard growth charts. Familial (or constitutional) tall stature is the most common cause; children are born tall to tall parents, and continue to grow at a normal rate in keeping with their mid‑parental height expectation.27
Endocrine causes of tall stature are less common but important to consider. Precocious puberty (<8 years of age for girls, <9 years of age for boys), congenital adrenal hyperplasia and hyperthyroidism can all present with rapid growth and advanced bone age. If missed, such conditions can lead to irreversible early closure of epiphyseal growth plates and significantly attenuated final adult height. Therefore, these causes warrant urgent referral to a paediatric endocrinologist for investigation and management. Rare causes include GH excess, such as GH-secreting pituitary adenomas and pituitary gigantism.
Obesity may lead to early adrenarche and increased linear growth. This temporary increase in height is counterbalanced by earlier onset of puberty by 0.6 years for girls and 0.7 years for boys and, hence, early closure of epiphyseal growth plates.28 Therefore, final height is consistent with their genetic potential.
Chromosomal and syndromic causes of tall stature also need to be considered. With a prevalence of one in 450, Klinefelter syndrome (47 XXY) is the most common aneuploidy with tall stature.29 Other syndromal causes to consider include homocystinuria and Marfan syndrome, which present with similar phenotypic features, and rare overgrowth syndromes such as Sotos syndrome.27,30
Referring to specialist service
When and whom to refer to specialist paediatric services:
- Height less than the first centile or significantly out of keeping with mid-parental height expectation.
- Growth velocity <25th centile (for bone age or level of maturation).
- Height crossing two centile lines on the growth chart (this is highly suspicious of a pathological diagnosis).11
- Delayed puberty, as defined above.
Consider general paediatric medicine or appropriate specialist paediatric referral if a non-endocrine cause is identified.
Investigations to consider prior to referral
Simple blood tests to exclude non-endocrine causes of poor growth should be considered first, including a full blood count, urea and electrolytes, liver function, calcium/magnesium/phosphate, erythrocyte sedimentation rate and coeliac serology. Simple endocrine tests should include thyroid function testing (TSH, FT4) and IGF-1. Other endocrine tests, such as blood GH and IGF binding protein 3 levels, are of limited utility. GH is pulsatile and, therefore, random levels are not diagnostic. Blood tests assessing pubertal status need to be correlated with clinical findings and are often unhelpful.
Karyotype testing should be performed on all girls whose height is less than the first centile or significantly out of keeping with their mid-parental height, or with pubertal delay, even if the classical clinical stigmata of Turner syndrome are not present.
A bone age X-ray of the left wrist will provide an assessment of skeletal maturity, which is compared with the Greulich–Pyle bone age standard atlas. Bone age reporting is best performed by a radiologist with experience in this field. It is also helpful for the specialist to be able to view the image to see if they concur with the reported bone age.
Conclusion
Disordered growth encompasses a wide range of differential diagnoses during adolescence. Short and tall stature in otherwise well children are most commonly due to inherited factors and warrant monitoring only. It is important to take a detailed history as the underlying cause of growth disturbance will often be elicited here. In addition, careful examination and simple baseline investigations will aid a clinician in eliminating diagnoses and enable clearer referral to general paediatric or paediatric endocrinology services as appropriate. Constitutional delay of puberty is a common presentation, especially for males; consideration should be given for referral to discuss pubertal induction if over 14.5 years of age.
Authors
Tashunka Taylor-Miller MBBS, Paediatric Endocrinology Fellow, Department of Endocrinology, Royal Children’s Hospital, Parkville, Vic. Tashunka.Taylor-Miller@rch.org.au
Peter J Simm MBBS, Paediatric Endocrinologist, Department of Endocrinology, Royal Children’s Hospital, Parkville, Vic
Competing interests: None.
Provenance and peer review: Commissioned, externally peer reviewed.