Case
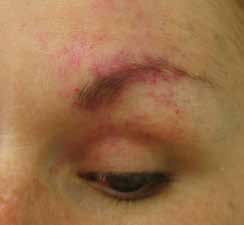 |
| Figure 1. Partially treated capillary malformation in left ophthalmic nerve distribution |
A woman aged 32 years presented seeking cosmetic improvement of a capillary malformation in the region of the ophthalmic division of her left trigeminal nerve. She reported that the capillary malformation had been present since birth. She had undergone investigation with computed tomography of the brain and no lesions were found. At the age of 7 years, she developed poor vision in her left eye and was found to have a partial retinal detachment due to a previously undiagnosed circumscribed ocular choroidal haemangioma. This was treated with argon laser photocoagulation, but progressed to complete retinal detachment and resulted in blindness in her left eye at the age of 13 years. One year later, she developed acute-onset glaucoma and underwent surgical intervention with lens extraction.
Despite the presence of ocular and cutaneous manifestations, she had never been diagnosed with Sturge-Weber syndrome (SWS).
Magnetic resonance imaging (MRI), performed when she was an adult, confirmed the lack of a leptomeningeal lesion and existence of a well-circumscribed left ocular choroidal haemangioma. She underwent cutaneous vascular laser treatment for cosmetic improvement of her capillary malformation. Figure 1 shows the capillary malformation in the left ophthalmic division of the trigeminal nerve after several cutaneous laser treatments.
Question 1
What is the inheritance pattern and incidence of capillary malformations and SWS?
Question 2
What are the clinical features of SWS?
Question 3
What investigations should be performed?
Question 4
How should this condition be managed?
Answer 1
A capillary malformation, or cutaneous port wine stain (PWS), is a common vascular malformation and is present in 0.3% of all newborn infants.1,2 Of all newborns presenting with a PWS, only approximately 3% have SWS.3,4 The likelihood of SWS is increased when the PWS manifests in the first (ophthalmic) or second (maxillary) division of the trigeminal nerve distribution.5
SWS is a rare, congenital vascular disorder. It is thought to be the result of a somatic mosaic mutation in the GNAQ gene, resulting in malformations in capillaries in fetal ectodermal tissues.1 The occurrence of SWS is sporadic and has no known causative risk factors.1,2 This rare and complex condition is still poorly understood. Endocrine disorders, such as growth hormone deficiency and central hypothyroidism, are also associated with this syndrome.1
Answer 2
SWS is a neurocutaneous disorder, classically presenting as a triad of a PWS in the area corresponding to the distribution of the trigeminal nerve, leptomeningeal angioma, and ocular choroidal malformation.1,6 The PWS may be unilateral or bilateral with one or more divisions of the trigeminal nerve being affected. When it is unilateral, it occurs on the side ipsilateral to the leptomeningeal angioma and choroidal malformation.1,2
However, the complete triad may not always be present and there is great variability in the clinical presentations of this syndrome.1,2,6 One classification method that has been proposed is the Roach scale:7
- Type I (PWS +/– leptomeningeal angioma +/– glaucoma)
- Type II (PWS +/– glaucoma)
- Type III (isolated leptomeningeal angioma).
SWS should be suspected if a patient presenting with a PWS has a co-existing ocular or neurological deficit. The capillary malformations of SWS should be differentiated from the proliferative lesions of infantile haemangiomas.
Leptomeningeal lesions occur most frequently in the parietal and occipital regions of the central nervous system.1,5 There may be associated hydrocephalus, hemianopia, contralateral hemiatrophy or hemiparesis, seizures and learning difficulties.1 These symptoms and signs are variable in severity and may be progressive. The variability of presentation is thought to be due to timing of the mutation during fetal development. The severity and progression of neurological deficits are determined by varying degrees of impaired blood flow.1
Ocular choroidal vascular lesions can be either diffuse or circumscribed and cause glaucoma, visual field defects and blindness. Other features signalling ocular involvement are heterochromia of the iris and angiomatosis of the sclera, conjunctivae or iris.1,6
Less frequently, involvement of the oropharyngeal, nasal and dental structures may occur. Rarely, there may be macrocephaly or facial hemi-hypertrophy.8
Answer 3
MRI with gadolinium contrast is the preferred imaging modality and is used to detect lesions of the ocular choroid and leptomeningeal structures.9 A negative early scan does not rule out SWS. Other investigations may include ocular ultrasonography, computed tomography and measurement of intraocular pressures.1
Once SWS is considered as a differential diagnosis, urgent referral for MRI and ophthalmological opinion should be undertaken.
Presentation as an adult, as in this case, highlights the possible sequelae of undiagnosed SWS. Ideally, SWS should be recognised in infancy or early childhood. However, this does not always occur and the diagnosis may be overlooked. One potential reason for this is that investigations that are now routine may not have been available when patients with SWS were children. The same applies in migrant populations where access to medical care in their native country may have been limited.
Answer 4
Early recognition and intervention may improve management and quality of life for patients with SWS.1 The aim of treatment is early and pre-emptive symptom control. Seizures are treated with anticonvulsants. Low-dose aspirin is considered to be beneficial.1 Cutaneous vascular laser therapy, such as pulsed dye laser (585–595 nm) is variably effective in treating PWS.1 Surgery may also have a role in the management of PWS.1 Treatment of ocular manifestations include photocoagulation therapy for choroidal vascular lesions, monitoring of intraocular pressures and pharmacotherapy or surgery for glaucoma.1 There is also a role for physical and cognitive therapy as this condition is associated with behavioural problems, learning difficulties and neurological deficits.1
SWS should be considered and investigated as is clinically appropriate in patients presenting with facial PWSs who have another neurological or ocular deficit. The clinician must have a high index of suspicion and needs to be familiar with the classical and partial forms of this condition. This case highlights the consequences of missed diagnosis, which equates to missed opportunities to minimise functional deficits that impact greatly on quality of life. Multidisciplinary or multimodal therapy may be required for ideal management of these patients.
Case follow-up
Our patient had excellent cosmetic improvement in her capillary malformation after treatment with a cutaneous vascular laser. She coped well with her visual impairment and glaucoma.
Competing interests: None.
Provenance and peer review: Not commissioned, externally peer reviewed.