Vascular birthmarks
When a patient presents with a vascular birthmark it is important to distinguish between a vascular tumour and a vascular malformation as this will predict outcomes and guide treatment. The Mulliken and Glowacki classification provides a framework to assist in differentiating vascular lesions (Table 1).1
Naevus simplex
Naevus simplex, colloquially known as 'salmon patch' and 'stork mark', are pale-pink to bright-red capillary vascular malformations with indistinct borders that blanch and become more prominent with crying and straining. Naevus simplex most commonly affects the forehead, glabella, upper eyelids and nape; most spontaneously disappear between the ages of 1 and 3 years.2 Naevus simplex are thought to be related to immaturity of the vasculature and have a prevalence of 20–60%. Treatment is usually not required.2
Naevus flammeus (port wine stain)
Naevus flammeus are capillary vascular malformations. They present as unilateral, homogenous red-to-violaceous macules involving the skin and, sometimes, the mucosa. They darken and thicken over time and have an incidence of 0.3%.3 Naevus flammeus can occur anywhere, but many involve the face where they follow the trigeminal nerve (fifth cranial nerve) distribution (branches 1–3 or V1–3)4 (Figure 1). While largely a cosmetic concern, a naevus flammeus on the face may be a marker for ophthalmologic or neurologic structural abnormalities and therefore further investigation is warranted.3
Sturge-Weber syndrome is defined as a facial naevus flammeus in association with an ipsilateral vascular malformation of the leptomeninges and eye.4 Location of the naevus helps identify newborns at risk of underlying abnormalities. If the opthalmic nerve (V1) territory is involved, an ophthalmological examination should be performed. If the V1 territory is associated with ophthalmologic or neurologic abnormalities, or if there is eyelid involvement or extension into the territories of maxillary (V2) and mandibular (V3) nerves, or if the naevus flammeus is bilateral, neuro-imaging should be organised with specialist review. Parents of the patient without V1 involvement may be reassured and informed about vascular laser as an option for treatment.3
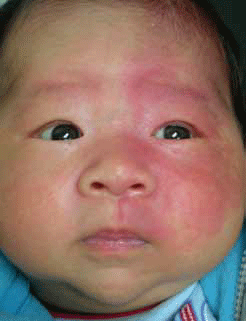
Figure 1. Naevus flammeus involving trigeminal nerve distribution V1 and V2
Table 1. Mulliken and Glowacki classification of vascular birthmarks1
| | Haemangioma | Vascular malformation |
|---|
| Histology |
Endothelial cell proliferation |
Rapid postnatal growth and slow involution |
| Histology |
40% are present at birth but usually appear as a small red mark |
Present but not always clinically apparent |
| Course |
Rapid postnatal growth and slow involution |
Grows in proportion with the person throughout their life |
| Female:male ratio |
3:1 |
1:1 |
Infantile haemangioma
Infantile haemangiomas (strawberry naevus) are benign vascular tumours. They occur in 5–10% of newborns, are more common in premature infants and have a female preponderance. Often unapparent at birth, they proliferate rapidly for several months, followed by gradual involution over several years.4–7 Superficial haemangiomas may be localised or segmental and appear as bright-red raised nodules (Figure 2). When located deeper in the dermis they appear as soft masses with a bluish tone. Segmental haemangiomas have a linear or geographic distribution and are more likely to have associated structural abnormalities.7
Most infantile haemangiomas involute without intervention and occupy uncomplicated sites. Those haemangiomas that are large or occupy the airway, eyes and vital organs can cause serious or life threatening complications and prompt specialist referral is required. Other complications include ulceration, secondary infection and scarring. Although resolution is considered complete in 76% of children by the age of 7 years, 50% will have minor residual changes such as telangiectasia or wrinkling.7
The treatment goal for infantile haemangioma is to prevent serious complications and permanent disfigurement (Figure 3). First line therapy has traditionally been systemic prednisolone during the proliferative phase at a dose of 2 mg/kg for 12–16 weeks. Side effects included irritability, cushingoid appearance, adrenal suppression and susceptibility to infection.7 In 2008, propranolol was noted to shrink a haemangioma when used to manage a child with hypertrophic cardiomyopathy.5 Since then it has emerged as a relatively safe and effective treatment for infantile haemangioma.6
However, currently there is no clearly defined guideline for the use of propranolol for haemangiomas. Most groups have used 1–3 mg/kg/day in divided doses after baseline assessments.6 Our current recommendations include assessment for the presence of bronchospasm, cardiac disease and vascular anomalies by a dermatologist and a paediatrician. Baseline investigations should include a blood sugar level, blood pressure, electrocardiogram and clinical photography.
Propranolol is commenced at 2 mg/kg/day in two divided doses for normal infants over 2 months of age. All patients should be closely monitored during the first dose for adverse cardiac and hypoglycaemic complications. This should be done in the hospital setting. Close follow up by a paediatrician or paediatric dermatologist is recommended. Treatment is often required until the child is 1 year of age, with subsequent slow weaning of dosage.8
Other treatments include topical and intralesional steroids, interferon alpha, imiquimod, vincristine, cyclophosphamide and excision, however, there is weak evidence to support these alternative treatments.
Pulse dye laser has been used with success, particularly in ulcerated haemangiomas.4
Epidermal naevi
Epidermal naevi are hamartomatous proliferations of the epithelium. They occur in one in 1000 live births with most appearing in the first year of life. Subtypes depend on the predominant cell type which include keratinocytes, the sebaceous gland, the pilosebaceous unit, and eccrine and apocrine glands.9 The naevi are composed of a localised clone of abnormal cells arising from a somatic mutation.4 Patients with unusual or distinctive keratinocytic naevi may need specialist assessment or genetic counselling.10
Naevus sebaceous (organoid naevus or naevus sebaceous of Jadassohn) are an epidermal naevus that present as well circumscribed plaques, which may be linear. Plaques are hairless, yellow and waxy and under the hormonal influences of puberty become more verrucous (wartlike). They appear most commonly on the scalp (Figure 4) but can present on the face and upper body. Benign and malignant tumours may arise in these naevi and a rapidly growing or ulcerated nodule within a sebaceous naevus should prompt excision. Previously, sebaceous naevi were excised in childhood to avoid the possibility of basal cell carcinoma development. As malignant progression in sebaceous naevi is rare, indications for surgery are controversial. Ongoing monitoring is a reasonable treatment option if there is no cosmetic impetus for surgery.4,10
Although epidermal naevi are most often sporadic, they can be part of a syndrome which includes neurologic, skeletal and ocular abnormalities, particularly if the naevus is extensive.
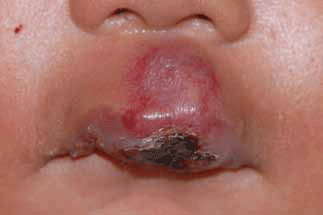
Figure 2. Ulcerated infantile haemangioma involving central upper lip before treatment
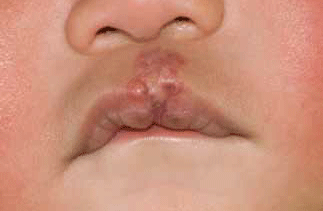
Figure 3. Infantile haemangioma involving central upper lip after treatment with prednisolone and propranolol for 4 months
Pigmented birthmarks
Dermal melanocytosis
Dermal melanocytosis (Mongolian spot) classically involves the lumbosacral area (Figure 5) and is seen at birth or soon after. It affects all races but has a higher prevalence in Asian children; most cases resolve in childhood.
The macular pigmentation of dermal melanocytosis has a blue-grey tone, usually involves less than 5% of the body, and is caused by sparse melanocytes residing in the mid to low dermis.
Site-specific dermal melanocytosis includes the naevus of Ota – a unilateral blue-brown facial patch often involving the ipsilateral sclera and occurring in dark-skinned people – and the naevus of Ito, which involves the upper back and shoulder. Both lesions persist throughout life and patients should undergo skin and eye surveillance due to the [rare] development of melanoma in these lesions. Naevus of Ota and naevus of Ito can be treated with pigment laser therapy.9
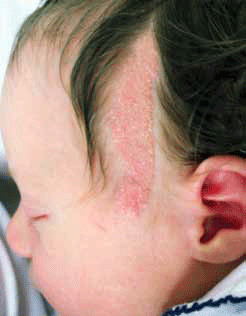
Figure 4. Linear naevus sebaceous involving left temple
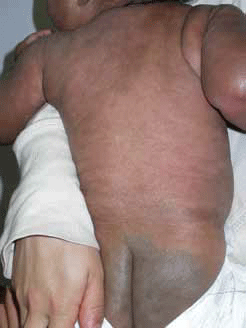
Figure 5. Dermal melanocytosis (Mongolian spot) demonstrating classic distribution over the lumbosacral area
Congenital melanocytic naevus
Congenital melanocytic naevi (CMN) consist of proliferations of benign melanocytes. They are present at birth (Figure 6) with an incidence of 1%.9 Naevi are classified according to their projected adult size: small (<1.5 cm), medium (1.6–19.9 cm) or large (>20 cm). Common acquired naevi, although similar in appearance to small CMN, appear later in childhood. These naevi are often flat, with colour variability initially, and evolve over time to become thicker with hypertrichosis and colour change.11 This natural evolution presents challenges in monitoring. Progress photographs are a valuable management tool.
The risk of developing melanoma in a CMN is significantly increased and there is a strong correlation with increasing size and the number of satellite lesions, with the highest risk occurring in large CMN >60 cm; these often develop in childhood.9,11 Patients can be reassured that the risk of melanomas in small and medium CMN is very low.
Neurocutaneous melanosis is a rare syndrome characterised by large or multiple CMN and tumours of melanocytes within the CNS that may be asymptomatic or cause neurological symptoms. Magnetic resonance imaging (MRI) may be considered in the diagnosis of high risk patients with extensive melanocytic naevi.11
Management of CMN focuses on identifying complications and optimising cosmetic outcome.11 Infants with large CMN should be assessed in a multidisciplinary setting within the first weeks of life. Uncomplicated small CMN carry similar risks to acquired naevi and may be observed in the primary care setting. Treatment options include serial observation, excision, dermabrasion, curettage, hair and pigment reducing lasers and chemical peels.12
Café au lait macules
Café au lait macules (CALM) are well circumscribed light to darkbrown macules. The macules can occur anywhere on the body and are usually noted in infancy and childhood. Their colour is due to increased melanin in keratinocytes rather than an absolute increase in melanocyte numbers.9 Solitary cALMs are common in up to 3% of healthy infants and 25% of children. Multiple cALMs are rare in healthy children and the majority of children with six or more cALMs will eventually be diagnosed with neurofibromatosis-1 (NF-1), potentially affecting multiple organ systems. children with three or more cALMs should be monitored for other features of NF-1.13 café au lait macules may also be seen in multiple other rare syndromes.13 Isolated cALMs may be treated with pigment laser therapy.9
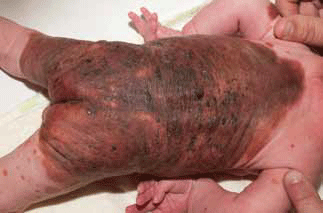
Figure 6. Large congenital melanocytic naevus with a truncal distribution and satellite naevi
Summary
Birthmarks are common and in most cases parents can be reassured that they are only of cosmetic significance and that for many children the appearance will improve over time. A minority of higher risk birthmarks have complications or systemic associations that need identification and further management.
Conflict of interest: none declared.
References