Infants with PWS are floppy and inactive, have a weak cry, a poor suck and feeding problems, which lead to growth failure. This is followed by several characteristic stages in which feeding problems diminish, weight rapidly increases and hyperphagia develops at 1–4 years of age.7,8 The pubertal growth spurt is generally absent and hence, short stature will be pronounced in adulthood.9 Scoliosis and kyphosis are common and may be severe.10 Hyperphagia, low muscle tone, respiratory and sleep problems, including excessive daytime sleepiness and lack of energy, exacerbate the tendency to become morbidly obese in the adolescent years, which is the major cause of mortality in adults with PWS.2,11
Growth hormone treatment
Subsidised growth hormone (GH) treatment has been approved by the Pharmaceutical Benefits Scheme for children with genetically confirmed PWS until the age of 18 years.12 Growth hormone treatment normalises height in children and improves body composition by reducing fat and increasing lean tissue mass in children and adults with PWS.13,14 The increase in muscle mass improves strength, tone and stamina for physical activity, which assists in weight management (Figure 1).15 Improvement in bone density with GH treatment may prevent the onset of osteoporosis, which commonly develops in adolescents and adults with PWS.16 The application for GH must be made by a paediatrician or endocrinologist and is only available on private prescription for adults. A preliminary sleep evaluation is obligatory if GH treatment is considered so that any existing respiratory problems can be addressed.

Figure 1. Two adolescent girls with Prader-Willi syndrome. One (left) received growth hormone treatment for 4 years; the other (right) received no growth hormone treatment
Incidence, prevalence and mortality
Information on the prevalence and mortality of people with PWS is limited. Mortality is high, approximately 3% per annum,17 and the average age of death was 33.2 years in a study of adults.18 In a recent Victorian study, however, the probability of survival was 94% until 20 years of age, but then declined rapidly, especially for those who are obese.11
The number of people in Australia with known PWS by year of birth, based on data from the Australian Prader-Willi syndrome database, is presented in Figure 2. At least 261 cases have been genetically confirmed and are seen in hospital-based PWS clinics. One in 17 000 would be a conservative estimate of the birth incidence in the past two decades. Another 79, mainly older individuals with PWS are known to the Prader-Willi Syndrome Association of Australia. Information on adults diagnosed with PWS is scarce and the total number of adult patients seen by specialists or general practitioners is currently unknown. Diagnosis of adults may be based on clinical characteristics only, which are not always confirmed genetically.
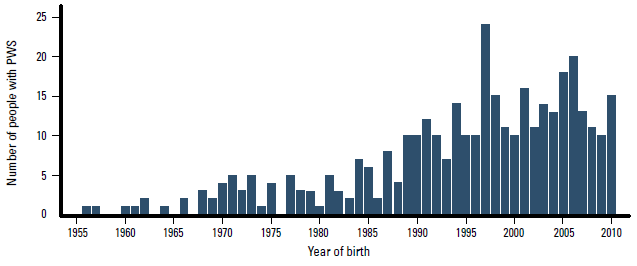
Figure 2. Number of people with Prader-Willi syndrome by year of birth in Australia since 1955
Data from the Australian Prader-Willi syndrome database and the Prader-Willi Syndrome Association of Australia
Genetic diagnosis
The diagnosis of PWS has improved dramatically since 1989 when genetic testing for the various subtypes of the condition became available. These tests detect lack of expression of paternally contributed genes on chromosome 15q11–q13 due to deletion or maternal uniparental disomy.19 DNA methylation analysis has a high reliability and specificity to test for all subtypes of PWS in contrast to previous diagnostic strategies, which relied on either clinical characteristics or chromosome studies.2
Diagnosis of children with PWS
Early diagnosis of PWS is important,20 as late diagnosis may result in heightened behavioural problems.21 Especially over the past decade, diagnosis of infants has improved their quality of life by multidisciplinary health management focusing on early intervention.22 All infants with characteristic signs of PWS, as described in Table 1, should be referred for genetic testing.23,24 Referral to a paediatric multidisciplinary PWS clinic is recommended for additional care. These are available in most capital cities in Australia.
Table 1. Presenting signs and symptoms suggesting referral for genetic testing for Prader-Willi syndrome
| Infants |
- Hypotonia
- Poor suck or feeding problems
- Weak cry
- Inactivity
Plus
|
| Adults |
- Short stature
- Obesity
- Mild to moderate intellectual disability
Plus
- Hypogonadism
- Hyperphagia
- Small hands and feet
- Thick, viscous saliva
Plus/minus
- History of hypotonia, poor suck, general feeding problems in infancy
|
Diagnosis of adults with PWS
Each year new diagnoses of PWS are made in patients aged in their 20s and 30s. Many people in this group seem to have previously been given an alternative diagnosis,20 commonly general intellectual disability, Asperger syndrome, autism spectrum disorder or even some other chromosomal abnormality, such as a PWS-like subtype of Fragile X syndrome.
The GP or allied health professional can play a major role in identifying adults with PWS by re-assessment of an existing clinical diagnosis. For example, if an adult patient is short, obese, has intellectual disability or learning difficulties, and also has hypogonadism, small hands and feet, insatiable appetite and thick, viscous saliva (Table 1), a re-investigation for PWS is warranted.20,23 In addition, a history of general feeding problems in early infancy that may have required tube feeding can often be considered confirmation of a clinical diagnosis of PWS (Table 1). Speech and articulation problems as well as skin picking are other highly characteristic features of PWS.24
Conversely, adult patients with a clinical diagnosis of PWS who do not have the characteristic features of hypogonadism and small hands and feet, mentioned in section 2 of Table 1, are likely to fail genetic confirmation of PWS.23 Indeed, 10 out of 56 individuals diagnosed with PWS from disability services records failed genetic confirmation in a Western Australian study.25
Management in general practice
While PWS is not common, patients with PWS visit their GP frequently due to the numerous chronic and sometimes acute problems related to obesity, respiratory function, sleep and behaviour. In addition, there are incidents such as choking, falls and injuries due to poor tone, coordination or intellectual disability. Patients with PWS are often hospitalised or need referral to specialist care. Table 2 provides an overview of common problems encountered or followed up in general practice.20,26–28
Table 2. Problems in Prader-Willi syndrome commonly encountered in general practice23,26–29
| System | Common problems |
|---|
| Respiratory |
Snoring
Sleep apnoea and hypopnoea
Excessive daytime sleepiness
Infections (eg. pneumonia)
Asthma |
| Endocrine |
Type 2 diabetes mellitis
Hypothyroidism |
| Cardiovascular |
Hypertension
Chronic leg oedema |
| Gastrointestinal |
Constipation
Reflux
Inguinal hernia |
| Musculoskeletal |
Fractures
Dislocations
Tendon tears
Heavy bruising
Osteoporosis |
| Dermatological |
Infection due to trauma from skin picking
Erysipelas |
| Psychological |
Behavioural issues
Psychosis
Sexuality/abuse
Difficulties with self-care/hygiene |
Caution in interpretation of seemingly minor complaints in PWS
Often the severity of a clinical symptom may be underestimated until the condition warrants immediate attention. The following advice is specific to patients with genetically proven PWS.29
High pain threshold
Minor fractures or injuries are difficult to diagnose when there are few or no complaints of pain. Often it may be some days before a problem is suspected (eg. when a bruise or swelling doesn’t disappear). A referral for an X-ray is advised whenever a fall has preceded a minor complaint of a sore limb.
Thermoregulation problems – lack of fever
Patients with PWS may not develop any fever, despite having a serious infection. Patients with PWS are especially prone to respiratory infections such as pneumonia. Undiagnosed, this can lead to death. On the other hand, ‘sudden excessive fever’ also occurs and may lead to fits. Both hyper- and hypo-thermia may occur in extreme weather conditions due to problems in thermoregulation.
Stomach problems
Overeating may lead to overextension and even rupture of the stomach. Due to high pain tolerance and difficulty in localising pain, the patient may just feel generally unwell. A referral to an emergency department is essential if the person complains of general abdominal pain and a bloating of the abdomen, especially if binge eating may have occurred and the person is not obese. An X-ray or CT scan should be taken or, in cases of serious pain or fever, an ultrasound or endoscopy should be performed as acute gastric dilation, inflammation, gastroparesis and necrosis are all more common in PWS than in the general population.
Lack of vomiting
People with PWS rarely vomit. This may be a concern, for example, if uncooked, spoiled or raw pet food has been eaten or non-food items have been ingested. Treatment for food poisoning or intoxication, for example from medication, is advised. Emetics are generally ineffective and their use is not recommended. Pumping the stomach and the use of activated medical charcoal are advised. On the other hand, if vomiting does occur in a patient who rarely, if ever, vomits, this may be a sign of a very serious or life-threatening problem.
Adverse reactions to medication or anaesthesia
Unusual reactions to standard dosages of medications or prolonged responses to sedation have been recorded.
Key points
- Prader-Willi syndrome is a severely disabling genetic condition of short stature and obesity.
- Growth hormone treatment has been endorsed to improve height and body composition of people with genetically confirmed PWS until the age of 18 years.
- Complications of obesity are a major cause of morbidity and early death in adults with PWS.
- A diagnosis of PWS in adults is often missed. In addition, diagnosis of PWS on only clinical characteristics is uncertain.
- A high pain threshold, absence of fever or vomiting, adverse reactions to medication or anaesthesia are specific concerns in patients with confirmed PWS, therefore genetic diagnosis is important.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Acknowledgements
The author is grateful to Dr Mieke van Driel, Professor of General Practice, for reviewing the manuscript and the Primary Health Care Research Evaluation and Development (PHCRED) program of Bond University, which stimulated her interest in primary care. The author acknowledges the contribution of the Australian Prader-Willi syndrome database and the Prader-Willi Syndrome Association of Australia regarding the number of people with PWS. The author is a recipient of a 2008 Pfizer Investigator Initiated Research Grant for instigating the Australian Prader-Willi syndrome database.